GROMACS version: 2025.3
GROMACS modification: Yes/No
Here post your question:
Hello Gromacs Forum:),
towards the above topic, I am using the NSF Slurm Cluster to execute the below commands:
gmx grompp -f inputs/nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr
gmx mdrun -deffnm nvt
to execute these the Slurm Command is:
$ sbatch gromacs.sh
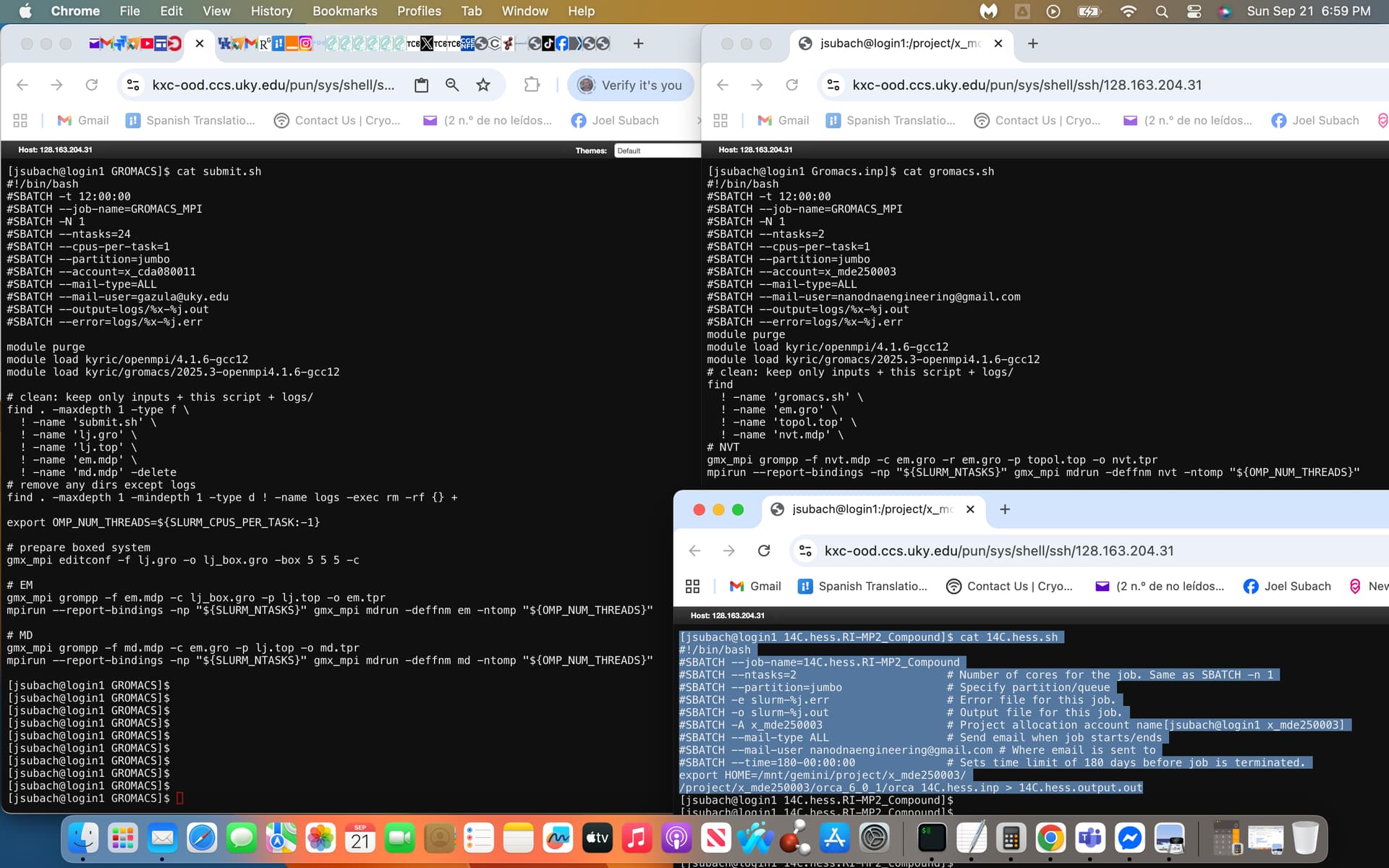
with gromacs.sh being the script file that I had modified from the sample submit.sh script file provided by NSF, attached is a screenshot of the former on the right and later on the left with an additional .sh script called 14C.hess.sh that has currently been successfully executed within Slurm exhibited on the bottom right highlighted in light blue.
I used the sample script (submit.sh) and the currently running script (14C.hess.sh) to modify the gromacs.sh script exhibited within the screenshot.
And these were the commands I executed to download the Gromacs on the Slurm:
module purge
module load kyric/openmpi/4.1.6-gcc12
module load kyric/gromacs/2025.3-openmpi4.1.6-gcc12
module list
gmx_mpi -version
This may be beyond the scope of the Gromacs Forum and may instead be best answered via the NSF Admin, however, if any users are experienced with success towards modifying my gromacs.sh script towards successful execution feel free to answer (I had executed as is and it failed), thanks:)
What happened when it failed? Without an error message, we’re only guessing. The best option is to work with the cluster’s admin to troubleshoot.
Hi Justin thank you for your kind update:)
Subsequent to execution via:
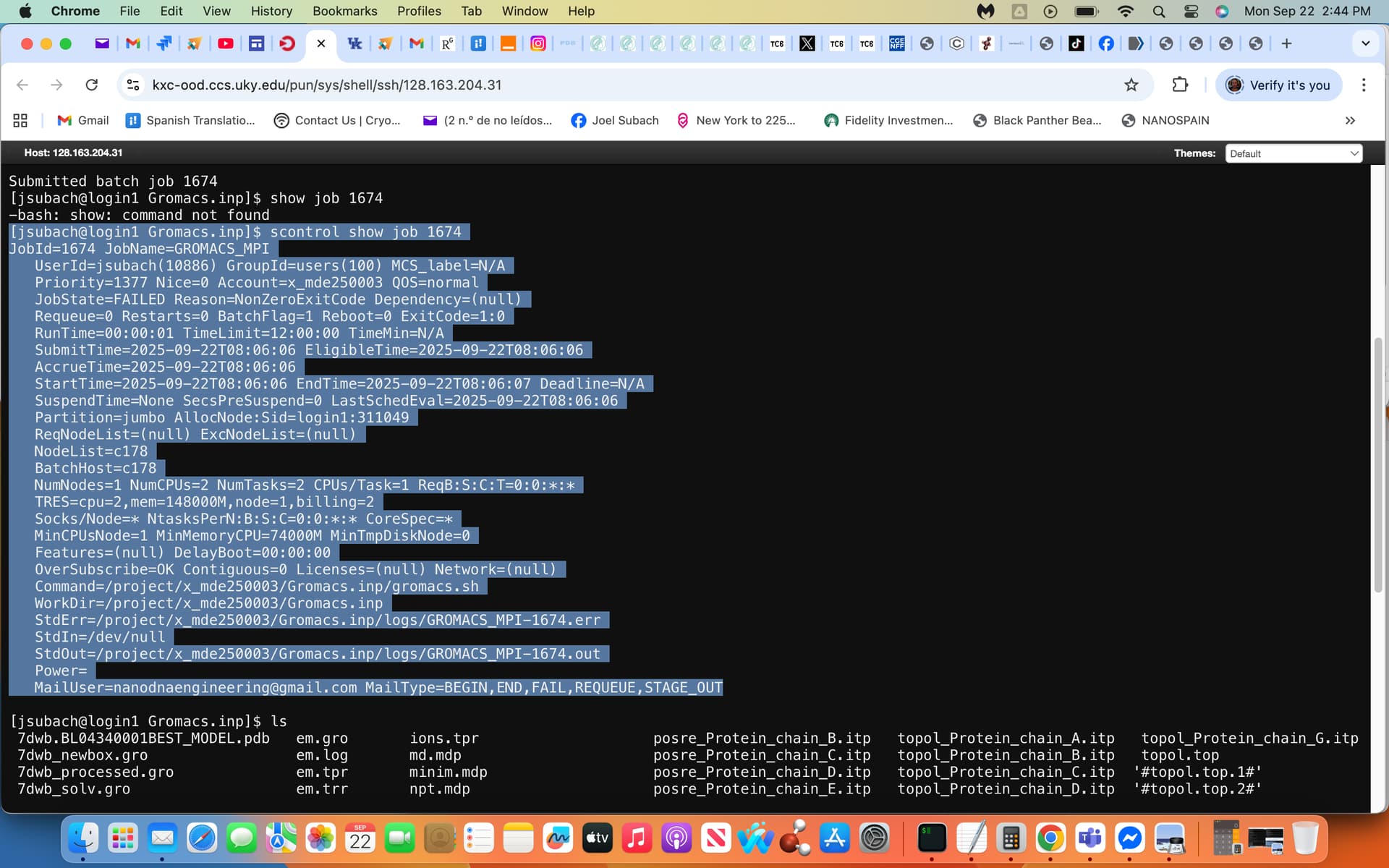
sbatch gromacs.sh, no files were generated i.e. no output.out file etc.
via inputting scontrol show job 1674 the following output failure generated the attached screenshot, yes I agree I will inquire further with the Slurm Admin. exhibiting this screenshot as well.
Hello Gromacs Forum, the solution to the above problem is below and attached (see light-blue highlighted .sh Script) via the NSF Admin:
Hi Joel,
I have a working script stored as
/mnt/gemini/project/x_mde250003/Gromacs.inp/gromacs_corrected.sh
There were a couple of issues in your gromacs.sh. It was missing the line that begins with “export OMP…”
I also deleted the lines that run “find”. I think the purpose of these is to clean up unwanted files in case you wanted to rerun the job. The edits you made to the find command likely would have caused errors.
I submitted the job using the corrected script and saw that it was running for a few minutes, and it did not get the error that you showed. I canceled it because I saw it was working. Go ahead and run the corrected script through sbatch (just change the top lines pertaining to the account name, job name, etc., as it was run under my account previously).
Feel free to inquire further and Good Luck!:)