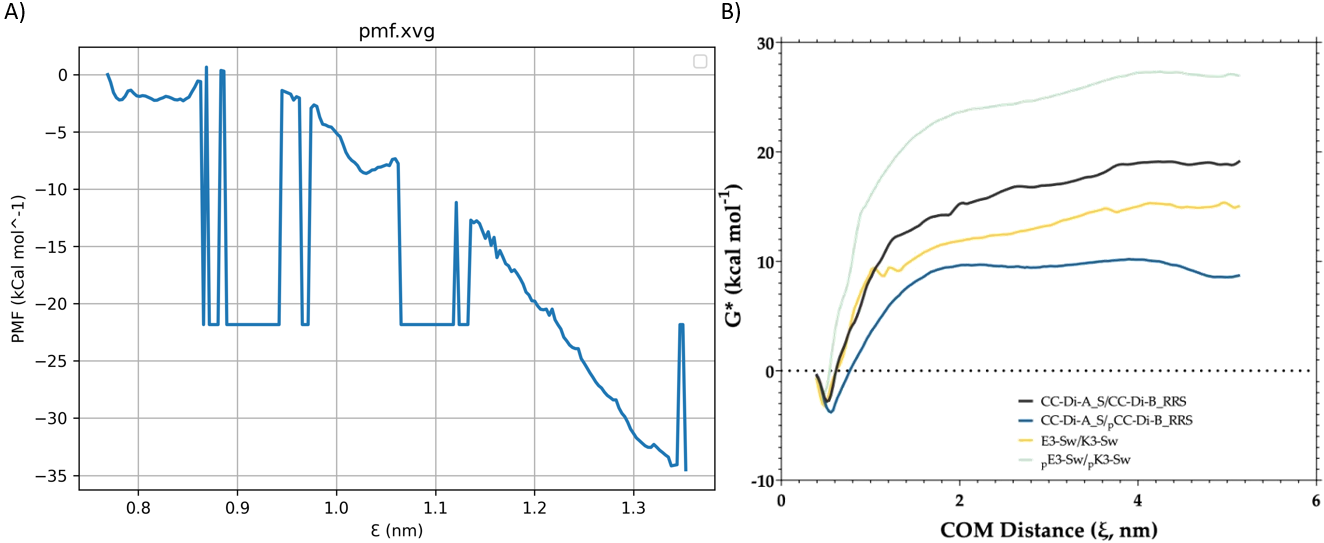
Hello!
I am using the tutorial as a guide but using a different system. I have two alpha-helix structures each 35 residues in length which I identified as Chain_A and Chain_B. The goal my project is to perform umbrella sampling to determine the potential of mean force (PMF) or free energy (G) in my peptides. To generate the 501 conf*.gro files (; Pull code snippet from md_pull.mdp), I ran the steered MD simulation and extracted the frames.
; Pull code
pull = yes
pull_ncoords = 1 ; Only one reaction coordinate
pull_ngroups = 2 ; Two groups defining one reaction coordinate
pull_group1_name = Chain_A
pull_group2_name = Chain_B
pull_coord1_type = umbrella ; Harmonic potential
pull_coord1_geometry = distance ; Simple distance increase
pull_coord1_dim = N N Y
pull_coord1_groups = 1 2
pull_coord1_start = yes ; Define initial COM distance > 0
pull_coord1_rate = 0.01 ; 0.01 nm per ps = 10 nm per ns
pull_coord1_k = 1000 ; kJ mol^-1 nm^-2
From the tutorial, I used the bash script to create the summary_distances.dat file, however there were no two configuration files with a spacing of 0.2 nm. As per the tutorial, I ran an NPT equilibration (npt_umbrella.mdp snippet below) with commands like:
gmx grompp -f npt_umbrella.mdp -c conf6.gro -p topol.top -r conf6.gro -n index.ndx -o npt0.tpr
gmx mdrun -deffnm npt0
…
gmx grompp -f npt_umbrella.mdp -c conf486.gro -p topol.top -r conf500.gro -n index.ndx -o npt24.tpr
gmx mdrun -deffnm npt24
; Pull code
pull = yes
pull_ncoords = 1 ; Only one reaction coordinate
pull_ngroups = 2 ; Two groups defining one reaction coordinate
pull_group1_name = Chain_A
pull_group2_name = Chain_B
pull_coord1_type = umbrella ; Harmonic potential
pull_coord1_geometry = distance ; Simple distance increase
pull_coord1_dim = N N Y
pull_coord1_groups = 1 2
pull_coord1_start = yes ; Define initial COM distance > 0
pull_coord1_rate = 0.0 ; Restrain in place
pull_coord1_k = 1000 ; kJ mol^-1 nm^-2
Following this, was the umbrella sampling (md_umbrella.mdp snippet below) with commands like:
gmx grompp -f md_umbrella.mdp -c npt0.gro -t npt0.cpt -p topol.top -r npt0.gro -n index.ndx -o umbrella0.tpr
gmx mdrun -deffnm umbrella0
…
gmx grompp -f md_umbrella.mdp -c npt24.gro -t npt24.cpt -p topol.top -r npt24.gro -n index.ndx -o umbrella24.tpr
gmx mdrun -deffnm umbrella24
; Pull code
pull = yes
pull_ncoords = 1 ; Only one reaction coordinate
pull_ngroups = 2 ; Two groups defining one reaction coordinate
pull_group1_name = Chain_A
pull_group2_name = Chain_B
pull_coord1_type = umbrella ; Harmonic potential
pull_coord1_geometry = distance ; Simple distance increase
pull_coord1_dim = N N Y
pull_coord1_groups = 1 2
pull_coord1_start = yes ; Define initial COM distance > 0
pull_coord1_rate = 0.0 ; Restraint in place
pull_coord1_k = 1000 ; kJ mol^-1 nm^-2
Lastly, I concatenated the respective files and ran a WHAM analysis:
ls umbrella*.tpr | sort -V > tpr-files.dat
ls umbrella*_pullf.xvg | sort -V > pullf-files.dat
gmx wham -it tpr-files.dat -if pullf-files.dat -o pmf.xvg -hist histogram.xvg -unit kCal
Thanks again for your help, I’ve been stuck for some time now