Hello all,
I have a question about umbrella sampling in Justin’s tutorial.
In the umbrella analysis section, the WHAM machenism derives the PMF.
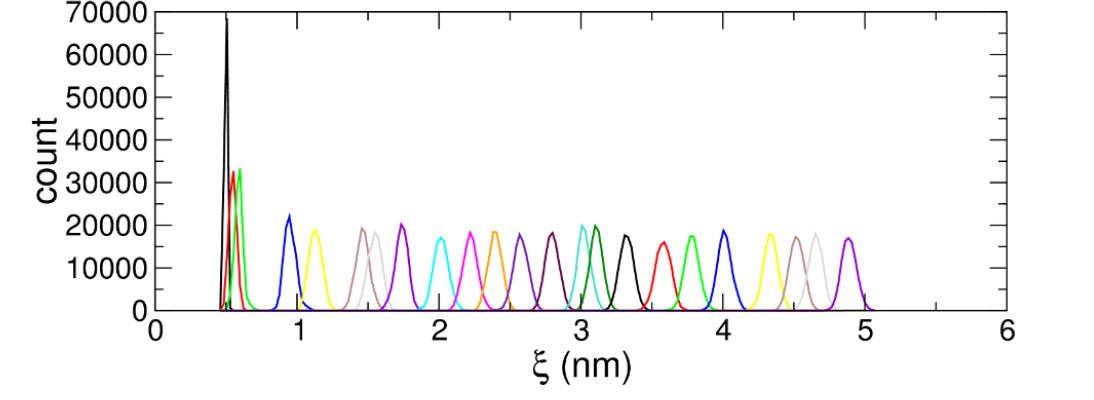
I have a question about one of the graph showing the umbrella sampling process.
I konw this graph is actually a curve averaged from the histogram and my question is does each column of the histogram represent peptide A of the same 3d structure or peptide A with the same distance from the COM of peptide A to the center of the sampling window?
Thank you.
Jiang
The X axis is the position along the reaction coordinate, i.e., the pull distance you have specified. In your case, that sounds like it would be distance of COM of peptide A to the reference position in your pull settings. A large number of peptide configurations can be represented at a single point along the reaction coordinate.
Hi MagnusL, thank you for your explanation. So my following-up question is if the column contain peptides with similar distance between COM to the reference (the window center) , does it mean peptides fall into the same column have similar configurations (or structures, or energy state)? Why here the the distance from the COM to the reference point could be used to represent the state of the peptide? For large and more complex proteins, does similar rule still apply?
You are using umbrella sampling along a pull coordinate. You decide what the reference should be (in the .mdp file). You can make the pull coordinate/reaction coordinate more or less complex.
But if you are not interested in the position along the reaction coordinate you should probably use another technique than umbrella sampling.
Thank you for your explanation. Yes the pull coordinate is simply the distance between the COM of the peptide A to that of the peptide B (the reference). For umbrella sampling, I chose a series of points along the pull coordinate with a fixed distance from each other. During an umbrella sampling, the peptide A is held at the chosen point by the umbrella force but should deviate from the chosen point during an simulation. But my hunch tells me there should be different configurations of peptide A that have similar or even the same deviation from the chosen point. Why the analysis algorithm seems not take this into consideration and simply use the probability of distance to calculate the free energy profile?
Yes, that is exactly what umbrella sampling is doing. It’s not taking your trajectory into account - only the pull coordinates (pullx.xvg or pullf.xvg files).
If you want to take different configurations into account you could, in principle, device a more complex reaction coordinate using pull-coord?-expression, see https://manual.gromacs.org/documentation/current/user-guide/mdp-options.html#mdp-pull-coord1-expression. You could also use multidimensional reaction coordinates, but then you should probably use other techniques than umbrella sampling.
I would propose that you first try to make a plan exactly what you want to study (and perhaps ask yourself why) and after that search for articles doing the same thing with MD simulations.
To follow up, during umbrella simulations you want to sample all configurations at each lambda point according to their probability distributions. But it’s only the position (using bins) along the reaction coordinate that is used to calculate the PMF.
In some cases, some configurations might not be accessible due to the umbrella restraints. Think of, e.g., a ligand tightly bound to a protein. There might be several configurations that would be possible for it to bind, but it would have to unbind to access those configurations. In such cases, umbrella sampling is not the best method. Then you would have more use for, e.g., AWH or metadynamics.
Hi MagnusL,
Thank you so much for your explanation. I’ve been haunted by this seemingly easy question and TBH, even though now I know the answer, I just try to memorize it but not be able to understand why. It’s like I know the equation of gravity but don’t know why it is that way. And thanks again for spending the time answering my questions. I didn’t have a major in physics and MD and my previous work and my insterests require me to learn MD. It’s so nice to have professionals like you here!
Best Regards,
Sincerely!
Jiang