GROMACS version:2021.5
GROMACS modification: No
Dear users,
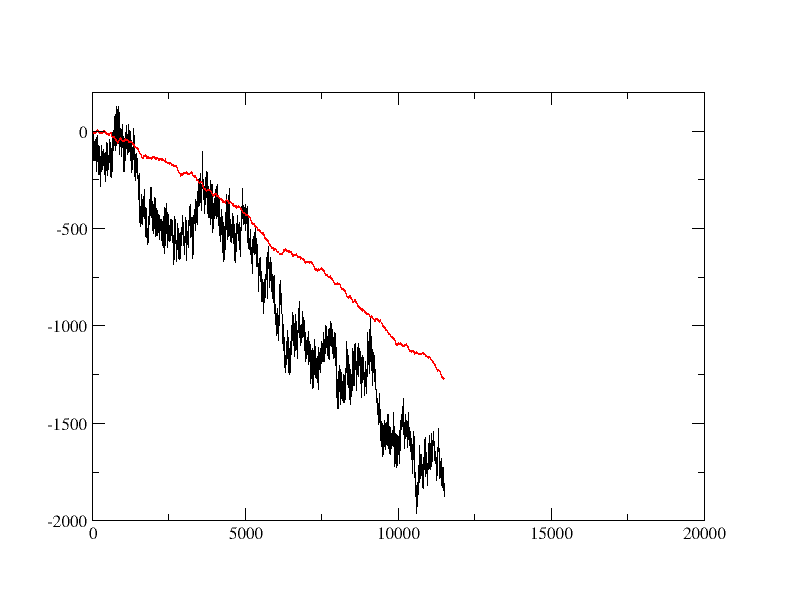
I am running an applied field simulations and facing issue with setting the center of mass removal. As I am doing mutational study on the protein, the initial runs were done using a position restraint of 100 on the CA atoms, later on the same simulation were performed without restraints and there was issues as the whole system started drifting upwards in Z direction(axis of electric field)(to be noted here the comm-mode=linear). This also had impact of current calculation and water permeation calculation(which reduced significantly). So should I use the center of mass removal or not, or should I just use COM removal for X,Y axis and not Z(direction of electric field), or is it okay to use the restraints?
For your reference the mdp parameters I am using is :
include
;define = -DPOSRES
; RUN CONTROL PARAMETERS
integrator = md
; Start time and timestep in ps
tinit = 0
dt = 0.002
nsteps = 25000000
; For exact run continuation or redoing part of a run
init-step = 0
; Part index is updated automatically on checkpointing (keeps files separate)
simulation-part = 1
; Multiple time-stepping
mts = no
; mode for center of mass motion removal
comm_mode = None
; number of steps for center of mass motion removal
nstcomm = 100
; group(s) for center of mass motion removal
comm_grps = SOLU_MEMB SOLV
; OPTIONS FOR WEAK COUPLING ALGORITHMS
; Temperature coupling
tcoupl = v-rescale
nsttcouple = -1
nh-chain-length = 10
print-nose-hoover-chain-variables = no
; Groups to couple separately
tc_grps = SOLU MEMB SOLV
; Time constant (ps) and reference temperature (K)
tau_t = 1.0 1.0 1.0
ref_t = 300 300 300
; pressure coupling
pcoupl = no
pcoupltype = semiisotropic
nstpcouple = -1
; Time constant (ps), compressibility (1/bar) and reference P (bar)
tau_p = 5.0
compressibility = 4.5e-5 4.5e-5
ref_p = 1.0 1.0
; Scaling of reference coordinates, No, All or COM
refcoord-scaling = No
; Electric fields
; Format for electric-field-x, etc. is: four real variables:
; amplitude (V/nm), frequency omega (1/ps), time for the pulse peak (ps),
; and sigma (ps) width of the pulse. Omega = 0 means static field,
; sigma = 0 means no pulse, leaving the field to be a cosine function.
electric-field-x = 0 0 0 0
electric-field-y = 0 0 0 0
electric-field-z = -0.0931 0 0 0
I would be very thankful for your help regarding the same!!