GROMACS version: 2023.3
GROMACS modification: No
Here post your question
The system contains:
-
3 single-pass membrane protein (∼3 k atoms)
-
400 truncated POPC lipids
-
2 000 DCLE tail-solvent molecules
-
TIP3P water + 0.15 M NaCl
what i did so far:gmx_mpi trjconv -s step7_1.tpr -f 200ns.xtc -pbc mol -center -o center.xtc -n index.ndx
Note that major changes are planned in future for trjconv, to improve usability and utility.
Will write xtc: Compressed trajectory (portable xdr format): xtc
Reading file step7_1.tpr, VERSION 2023.3 (single precision)
Reading file step7_1.tpr, VERSION 2023.3 (single precision)
Select group for centering
Group 0 ( SOLU) has 19071 elements
Group 1 ( MEMB) has 91324 elements
Group 2 ( SOLV) has 323925 elements
Group 3 ( SOLU_MEMB) has 110395 elements
Group 4 ( SYSTEM) has 434320 elements
Select a group: 1
Selected 1: ‘MEMB’
Select group for output
Group 0 ( SOLU) has 19071 elements
Group 1 ( MEMB) has 91324 elements
Group 2 ( SOLV) has 323925 elements
Group 3 ( SOLU_MEMB) has 110395 elements
Group 4 ( SYSTEM) has 434320 elements
Select a group: 3
Selected 3: ‘SOLU_MEMB’
Reading frame 0 time 0.000
Precision of 200ns.xtc is 0.001 (nm)
Using output precision of 0.001 (nm)
Back Off! I just backed up center.xtc to ./#center.xtc.1#
Last frame 2000 time 200000.000 → frame 1999 time 199900.000
→ frame 2000 time 200000.000
Last written: frame 2000 time 200000.000
then: gmx_mpi trjconv -s step7_1.tpr -f center.xtc -pbc whole -o whole.xtc -n index.ndx
:-) GROMACS - gmx trjconv, 2023.5-plumed_2.11.0_dev (-:
Executable: /usr/local/gromacs/bin/gmx_mpi
Data prefix: /usr/local/gromacs
Working dir: /home/nadia/smit/anm/w.omlac
Command line:
gmx_mpi trjconv -s step7_1.tpr -f center.xtc -pbc whole -o whole.xtc -n index.ndx
Note that major changes are planned in future for trjconv, to improve usability and utility.
Will write xtc: Compressed trajectory (portable xdr format): xtc
Reading file step7_1.tpr, VERSION 2023.3 (single precision)
Reading file step7_1.tpr, VERSION 2023.3 (single precision)
Select group for output
Group 0 ( SOLU) has 19071 elements
Group 1 ( MEMB) has 91324 elements
Group 2 ( SOLV) has 323925 elements
Group 3 ( SOLU_MEMB) has 110395 elements
Group 4 ( SYSTEM) has 434320 elements
Select a group: 3
Selected 3: ‘SOLU_MEMB’
Reading frame 0 time 0.000
Precision of center.xtc is 0.001 (nm)
Using output precision of 0.001 (nm)
Back Off! I just backed up whole.xtc to ./#whole.xtc.1#
Last frame 2000 time 200000.000 → frame 1999 time 199900.000
→ frame 2000 time 200000.000
Last written: frame 2000 time 200000.000
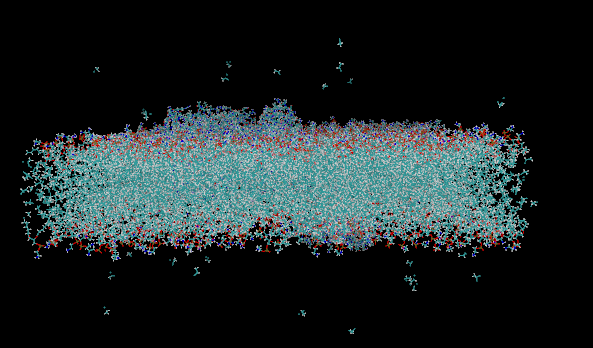
After step 2 the bilayer looks disintegrated: lipids are in pieces and DCLE clouds appear far above/below the slab (screenshot attached). Skipping -pbc nojump keeps the bilayer intact, but then individual DCLE molecules jump across the box edges frame-to-frame.
What I’m looking for
-
A robust command sequence (or FixBox settings) that
-
keeps each POPE lipid and each DCLE molecule whole,
-
centers the protein/bilayer in Z,
-
gives a continuous trajectory suitable for APL, RMSD, etc.
-