Permission to ask MD simulation where my input file preparation uses CHARMGUI where in my receptor complex 3BWM it is a type of bisubstrate protein, there are 2 ligands where one of the ligands is a ligand which has activity helping the work of the receptor (SAM) with the help of mg ions so that ions mg (metalloprotein) and SAM cannot be removed because they play a role in receptor activity, while compounds that bind to the active site which is the target of action of the drug must also be selected so that in charmgui when we choose protein, SAM, MG, and ligand (compound) it causes an error
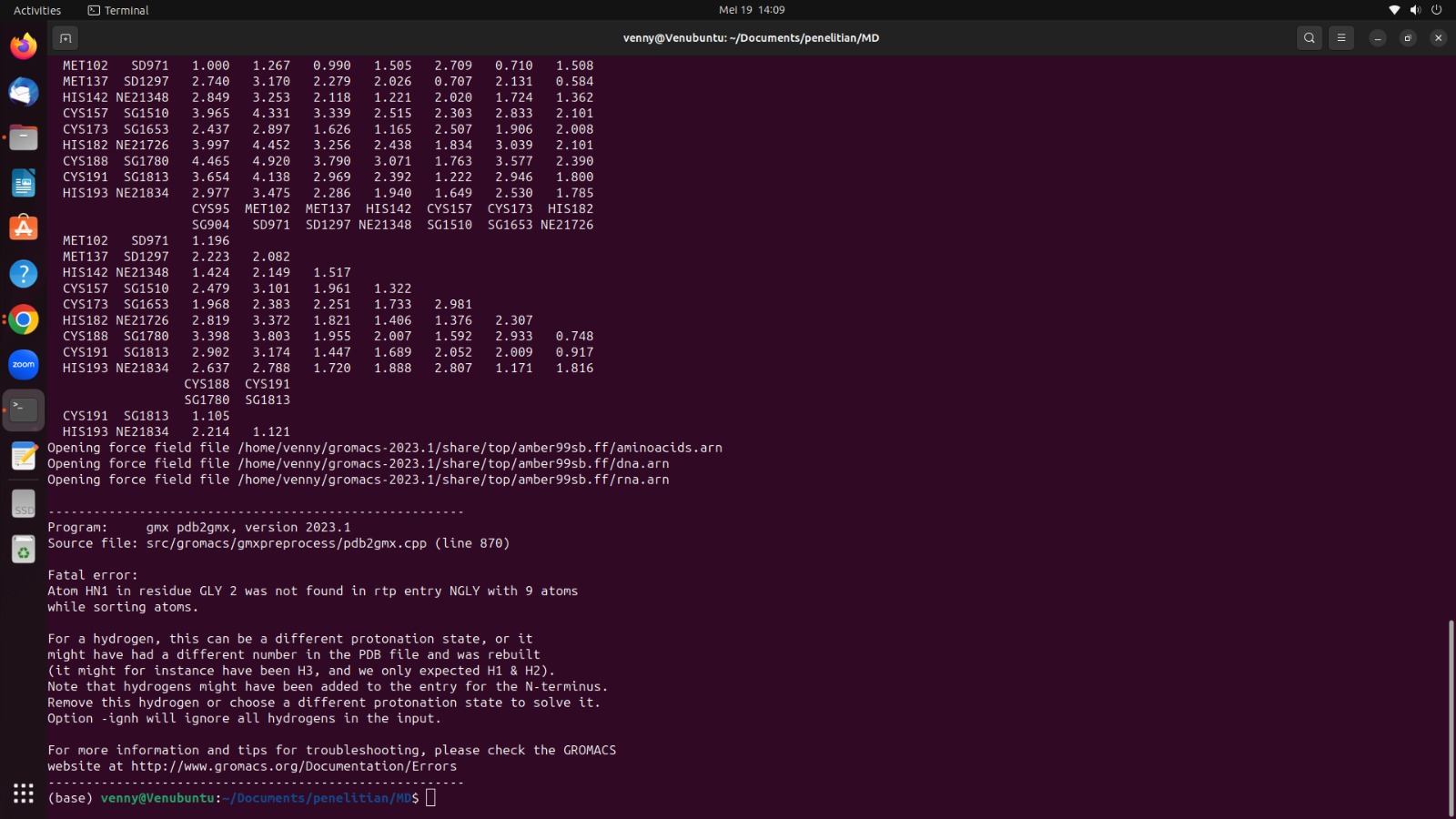
Then I also tried to make a protein topology manually according to the guide on the Gromacs tutorial simulation site and there was an error notification
permission to attach image:
please help me