GROMACS version:
GROMACS modification: Yes/No
Here post your question:
Hello I simulated a heptamer transmembrane protein 426-residues per chain (2982 residues total), using the methodology and indicated times (eg. 1-ns MD simulation) from Tutorial One: Lysozyme in Water. Would a recommendation of using 100-ns and any other time increases within this tutorial be warranted in this case towards confirming satisfactory RMSD etc. Analysis or considering the above the tutorial may be followed as is? Thanks if you know:), Joel
Refer to published studies of similarly sized systems for guidance, but large systems like you’re describing, including a membrane, often require hundreds of ns to even equilibrate before running data collection. RMSD doesn’t tell you anything about the adequacy of equilibration or sampling.
Hi Justin towards this thread, I will run a 1000 ns NVT Simulation on the NSF Cluster containing 1-node, 2-cpus and 74 GB of memory; is there a Time-Limit that you may recommend for this i.e. heptamer transmembrane protein 426-residues per chain (2982 residues total)? Thanks if you have any recommendations for these particular specs:)
There is no way to provide any kind of prediction. Your system will be huge (the number of protein residues is not the determining factor, it’s the total number of atoms) and the speed will depend on things like cutoffs and other algorithms. I certainly would never try such a simulation on 2 CPU cores. You need way more than that to achieve any kind of reasonable speed. Do some short benchmarking runs, a few thousand steps of MD, and you’ll have your answer.
…and can you please confirm that the below Gromacs Output towards my above thread is Mb Megabits in lieu of Megabytes? (The disk quota is limited to a few terabytes.). See-below:
WARNING: This run will generate roughly 9883504 Mb of data
starting mdrun 'Protein in water’500000000 steps, 1000000.0 ps.
slurmstepd: error: *** JOB 1677 ON c178 CANCELLED AT 2025-09-23T22:52:24 DUE TO TIME LIMIT ***[
jsubach@login1 Gromacs.inp]$
I could not confirm this from my search, thanks if you can:) and if you have an optimal core number feel free to recommend.
Hi Justin thank you for your kind update:) and if increasing the cores to 20 will subsequently decrease the roughly 9883504 Mb generated feel free to let me know (otherwise I will have to inquire with the NSF Admin. if there is a way that he can increase the few only terabytes to 10 or more in this case).
The output is not related to the number of cores you use for the simulation. It is a function of output options set in the .mdp file. For a large system, you probably can’t afford (in terms of space) to output to the trajectory as frequently as you’re specifying, and often adequate sampling requires far less frequent sampling than most people use.
Hi Justin and thank you:) I must then modify the Integration Time Step if I wish to keep the simulation time at 1000 ns? Feel free to recommend a modification to the below or anything else.
It would be helpful if you would post a complete .mdp file. The question is not about dt, that’s basically a standard setting. The large output volume is because your output settings (like nstxout-compressed, nst[xvf]out, etc.) are probably set too low and you’re writing out data much too frequently. If you’re writing to a .trr file, you probably don’t even need to be. Without seeing your full .mdp file, I’m just guessing, which is not productive in answering your question.
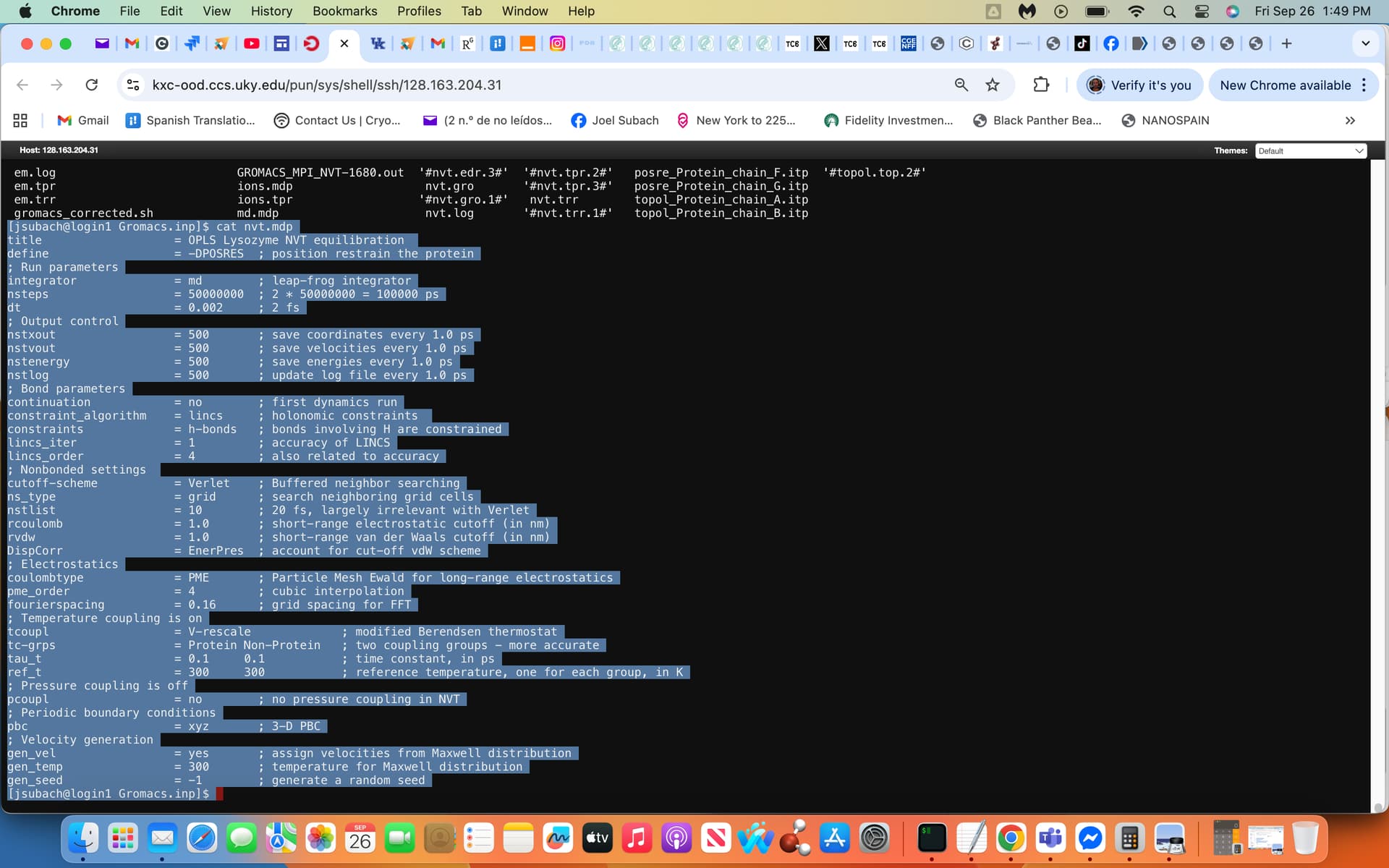
Hi Justin thank you:) and please view attached screenshot exhibiting in light blue the nvt.mdp file, currently the nsteps are set at 50,000,000 in this case 100 ns but prefer a 1000 ns simulation. I used the same nvt.mdp given in the tutorial with a single modification at the nsteps input. Thanks if you can provide me with any modifications of this file to allow a 1000 ns gmx mdrun -deffnm nvt while using a disk quota limited to a few terabytes.
You are saving high-precision coordinates and velocities every 1 ps, which is going to drive massive disk usage. Normally, compressed trajectory format (.xtc) is perfectly fine for standard MD, you don’t even need to write to the .trr file (nstxout and nstvout) and don’t save velocities unless you plan on using them for analysis (again, not something most standard MD uses). I recommend setting nstxout and nstvout to zero and then any other output (nstxout-compressed, nstenergy, and nstlog) to values no smaller than 5000.
and subsequent to your recommended above modifications to the nvt.mdp file the run has dropped from requiring 9,883,504 Mb to just 432 Mb of data for a 1,000 ns simulation.
The nvt.log file now exhibits the 1,250,000 step after 12-hours with 500,000,000 steps required. I am unsure if this run will finish in proportion to the above 12-hours i.e. 200-days to completion, accordingly if there then will be another/other modification to speed up completion feel free to comment, thanks again:)
Hi Justin concurrently I am executing a CHARMM-GUI generated Gromacs Equilibration, however, the CHARMM-GUI generated Gromacs Equalibration .mdp files are split into six-total steps for best equalibration considering the membrane influence. My Aim is 1000 ns per equalibration, accordingly would you suggest just splitting each of there .mdp files into 333 ns? Thanks if you have any input:)