GROMACS version: 2020.2
GROMACS modification: No
So I’m trying to run a tiny simulation on a very fast GPU, it’s 19,000 atoms on an Nvidia A100. I’m using the nvidia registries container NVIDIA NGC with the relevant flags. I have the relevant flags enabled (overkill actually as this is running on only one GPU, not several).
Here’s what I believe is true:
The whole simulation step was ported to GPU, except the neighbor list building. To me this means that the simulation can run on the GPU for nstlist number of steps in GPU memory, and only then is transferred back to the CPU for the neighborhood list to be built. [Or because a traj write out has been requested or similar].
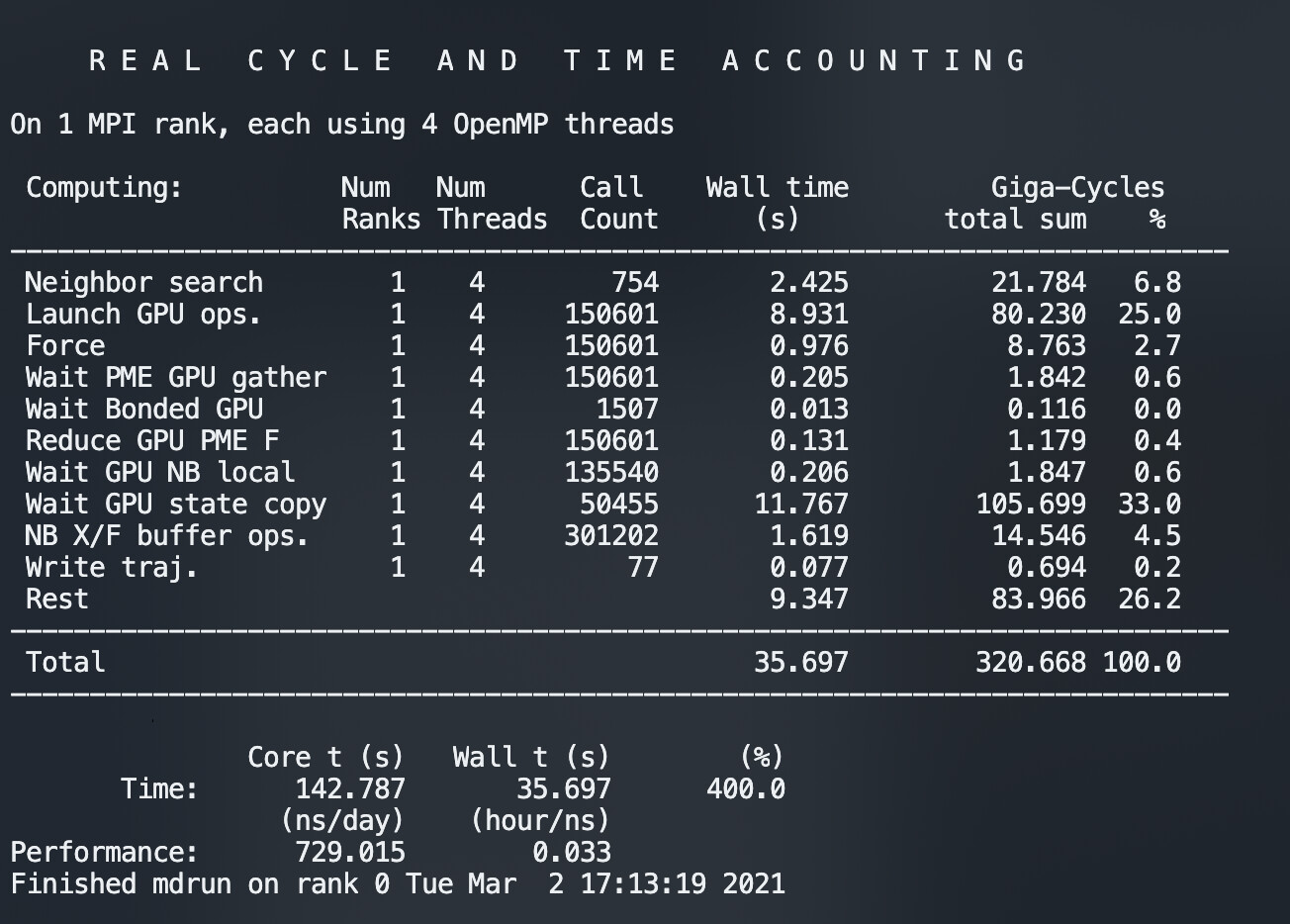
However what I see is that for the 150,601 steps (hence the number of force cycles calculated) the neighbor list is calculated just 754 times (yep, thats the nstlist 200) but the state is copied Wait GPU state copy 50,455 times, roughly every 3 steps. This is a huge drag. Why is it doing this?
Also if you have any tips on accelerating this (without bulking up the system size) it’d be much appreciated. I know GROMACS wasn’t designed for little systems and because the GPU is so fast these overheads pile up, but still.
Flags:
ENV GMX_GPU_DD_COMMS true
ENV GMX_GPU_PME_PP_COMMS true
ENV GMX_FORCE_UPDATE_DEFAULT_GPU true
Command:
gmx mdrun -deffnm md_prod -ntmpi 1 -ntomp 4 -nb gpu -bonded gpu -pme gpu -nstlist 200 -pin on -update gpu
Note: This is a 19,000 atom system with amber99sb-ildn and tip3p. It’s run here with just Justin Lemkuls classic tutorials options.
title = OPLS Lysozyme NPT equilibration
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 30000000 ; 2 * 500000 = 1000 ps (1 ns) so 60 ns
dt = 0.002 ; 2 fs
; Output control
nstxout = 0 ; suppress bulky .trr file by specifying
nstvout = 0 ; 0 for output frequency of nstxout,
nstfout = 0 ; nstvout, and nstfout
nstenergy = 2000 ; save energies every 10.0 ps
nstlog = 2000 ; update log file every 10.0 ps
nstxout-compressed = 2000 ; save compressed coordinates every 10.0 ps
compressed-x-grps = System ; save the whole system
; Bond parameters
continuation = yes ; Restarting after NPT
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds involving H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Neighborsearching
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet scheme
rcoulomb = 1.0 ; short-range electrostatic cutoff (in nm)
rvdw = 1.0 ; short-range van der Waals cutoff (in nm)
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling is on
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = Protein Non-Protein ; two coupling groups - more accurate
tau_t = 0.1 0.1 ; time constant, in ps
ref_t = 340 340 ; reference temperature, one for each group, in K
; Pressure coupling is on
pcoupl = Parrinello-Rahman ; Pressure coupling on in NPT
pcoupltype = isotropic ; uniform scaling of box vectors
tau_p = 2.0 ; time constant, in ps
ref_p = 1.0 ; reference pressure, in bar
compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Dispersion correction
DispCorr = EnerPres ; account for cut-off vdW scheme
; Velocity generation
gen_vel = no ; Velocity generation is off