So i wanted to use the membrane only system for building a bacterial membrane. I specified the lipid composition of the same in both Upper and Lower leaflets, all 6 steps ran and then I downloaded the tgz file, where i had 6.1 to 6.6 equilibration steps and a step7 which was the production step. after i run these steps one by one, my molecules are becoming too crowded. Any suggestions or help?
The default simulation length of “step7” in the CHARMM-GUI output is rather short. For a complex system, like a bacterial membrane, you would expect several hundreds of nanoseconds up to several microseconds of simulation time to obtain converged observables.
Again, what do you mean with “overlapping”? Are the atomic positions too close to each other and the simulation fails due to very high forces? Could you maybe post a snapshot of your system (e.g., from the last frame of your last simulation)?
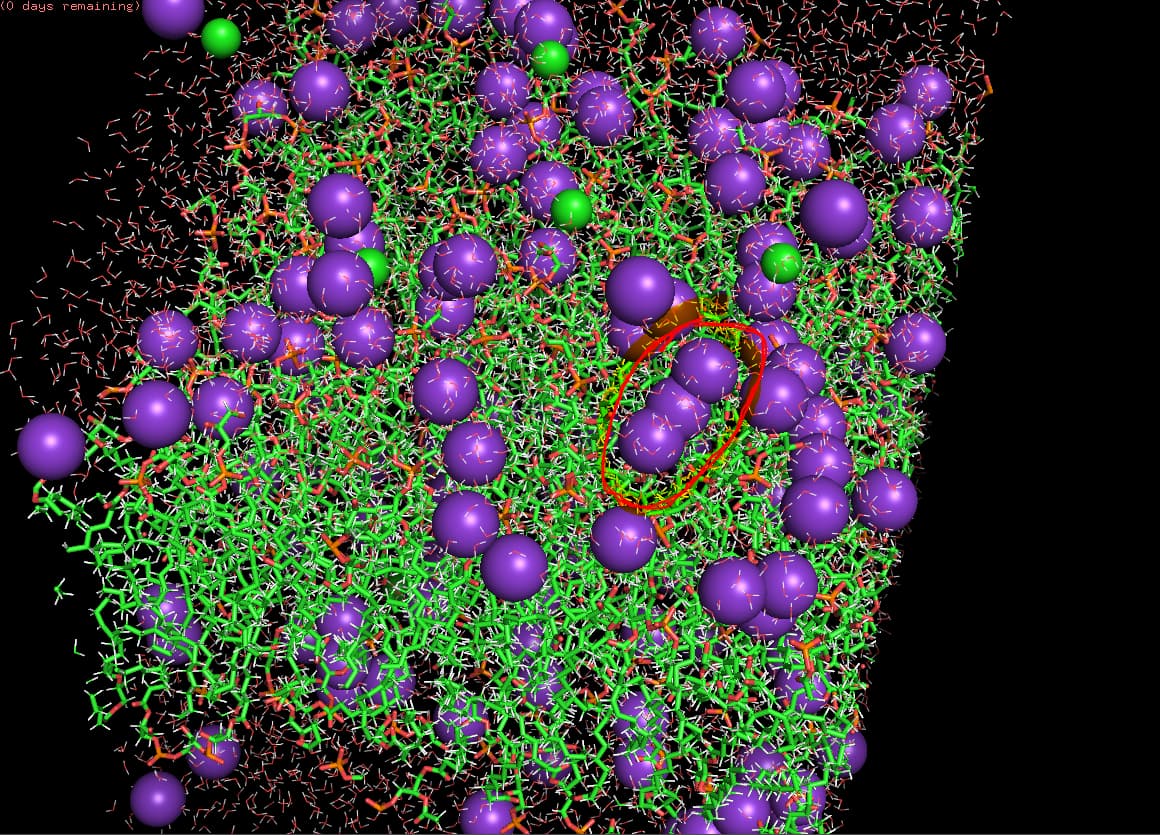
so I am adding a snapshot of a pdb file from my last trajectory. this is after production step by the way. I have highlighted in red circle where my molecules are closely packed and that is also present in other molecules, this is just one example. also, I checked if that could be an artifact but it actually appears to be joined.
the purple ones are cations like K⁺, Na⁺, or Ca²⁺ ions. Green spheres are anions. also I am trying to build a lipid membrane for gram positive bacteria. and then I want to embed my peptide later on. but yeah my doubt was how to have separate molecules for my membrane. also force field is charmm36
We need some more specifics, not cations “like” some list. Please tell us exactly what is in the system you’re showing us. This is important for understanding the proper functioning of the force field. Are you using all files directly from CHARMM-GUI? What is the lipid composition?
I had the same problems and I needed to generate the equilibration step nvt in CHARMMGUI, as output they give you the the input files to nvt equilibration and the results. I took the .pdb used as input to nvt which is without clashes