GROMACS version: 2020.4 patched with Plumed 2.7.0
GROMACS modification: Yes
Dear GMX users,
I am running coarse-grained well-tempered metadynamics (WT-METAD) on a membrane protein dimer using Elnedyn 2.2 (elastic network in combination with Martini force field) and I am facing problems with the box size mainly within the z-axis. This causes the membrane to look bizarre.
Here are the steps that I was following for the WT-METAD run:
1- Topology generation using Martinize.py
2- Box definition with gmx editconf
3- Insertion in the membrane with insane.py
4- Adding neutralizing counter ions.
5- Energy minimization
6- NVT equilibration with positions restraints on the protein backbone
7- NPT equilibration with positions restraints on the protein backbone
8- 1us unbiased MD run (no position restraints)
9- Extraction of the frame at 1us and starting the WT-METAD
The most important .mdp parameters that I am using are:
integrator = md
dt = 0.02
continuation = yes
cutoff-scheme = Verlet
nstlist = 20
ns_type = grid
pbc = xyz
verlet-buffer-tolerance = 0.005
coulombtype = reaction-field
rcoulomb = 1.1
epsilon_r = 15
epsilon_rf = 0
vdw_type = cutoff
vdw-modifier = Potential-shift-verlet
rvdw = 1.1
tcoupl = v-rescale
tc-grps = Protein_memb W_ION
tau_t = 1.0 1.0
ref_t = 315 315
Pcoupl = parrinello-rahman
Pcoupltype = semiisotropic
tau_p = 12.0
compressibility = 3e-4 3e-4
ref_p = 1.0 1.0
gen_vel = no
gen_temp = 315
gen_seed = -1
constraints = none
constraint_algorithm = Lincs
nstcomm = 100
comm-grps = Protein_memb W_ION
Regarding the WT_METD parameters, this is what I am using:
restr: UPPER_WALLS ARG=d AT=6.5 KAPPA=10000
METAD …
ARG=d,c
SIGMA=0.01,10
HEIGHT=1.2
PACE=500
BIASFACTOR=20
TEMP=315
LABEL=metad
CALC_RCT
… METAD
Here is an illustration of my problem -
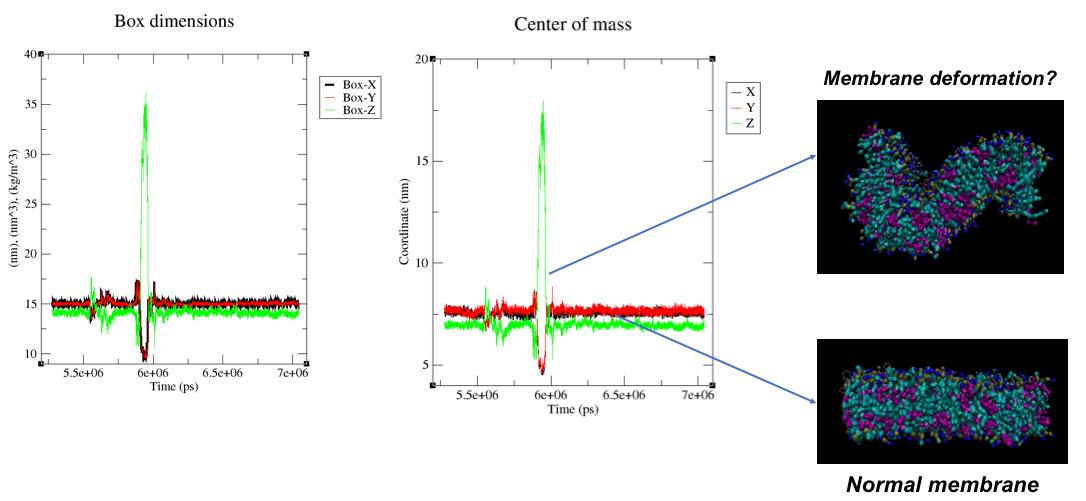
I included both box dimensions in the x, y, and z dimensions and also COM coordinates of the bilayer in time, and a visualization of the membrane.
My simulation is running in an NPT ensemble so it is normal to have some fluctuations in the box dimensions but in my case, the drift is very important and causes a wired behavior in the membrane shape. I guess this is not normal right?
Do you have any idea why there is such “isolated” drift in the box dimensions? Are these changes normal or acceptable? I used the recommended parameters for the Martini force field in the .mdp file but maybe something is not adapted for my system and needs to be adjusted?
I did some tests by coupling separately (pressure and temperature) the membrane, protein, and the solvent but still the same problems.
For the values of sigma in the WT-METAD, they are half the fluctuations of my CVs from my unbiased run (as recommended). The rest of the parameters seems acceptable to me.
Any help and ideas would be very appreciated.