GROMACS version: Gromacs 2019.1
GROMACS modification: No
Hello users,
Hello Justin,
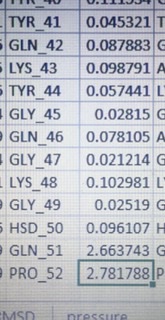
Had some doubt about gmx rms command, I was calculating rmsd for individual amino acids of peptide, in my case the rmsd for tail amino acids is shooting 100 fold. Command is correct, I cross checked, I am making structure whole before running rms