GROMACS version: 2020.1-Ubuntu-2020.1-1
GROMACS modification: No
Hi everyone!
I have some problem with system optimization.
I have a task to trace the molecular dynamics of berberine in water. To do this, I need to put berberine molecules in a water box. There was a problem described in the screenshot.
Used the command: gmx pdb2gmx -f *** -o *** -water spce
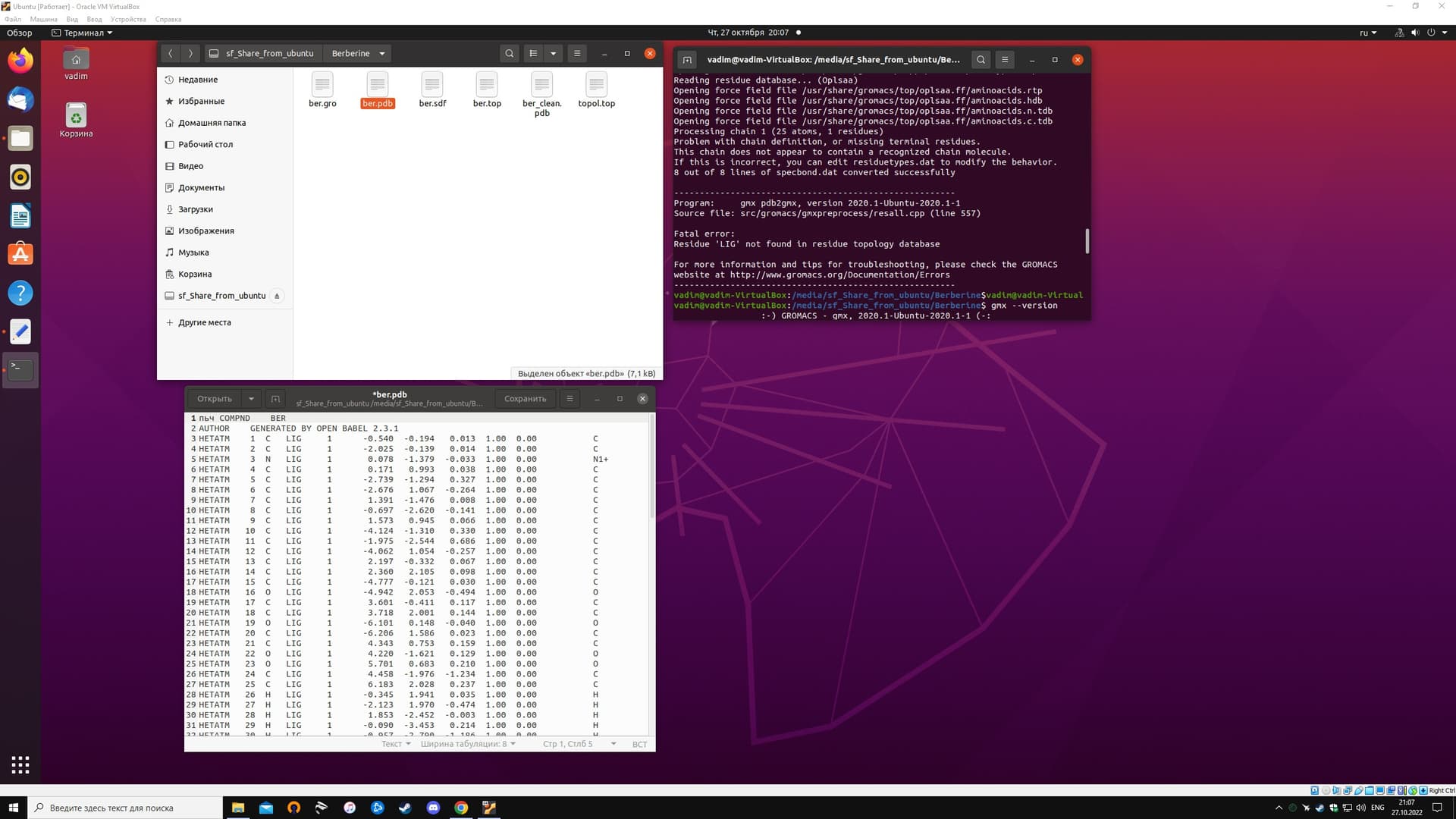
I know that GROMACS have a force fields for proteins. Is there an option to add a force field for non-protein structures?
There is no force field that will automatically parametrize any arbitrary species. You have to parametrize this molecule and provide a residue definition for it (.rtp entry) or use non-GROMACS tools to produce its topology.
Thanks a lot.
What a program i can use to get a .top file?
There’s one for every force field - CGenFF for CHARMM, Antechamber/AcPype for AMBER, LigParGen for OPLS, ATB for GROMOS, etc.
Thanks.
I will try to do something with this.
Forgot to ask. How can i get a LigParGen or something else? Is it addons for GROMACS?
With developing force field parameters for your molecule I’d suggest having a good look at the AMBER-DYES and CHARMM-DYES parameters and papers. This sort of molecule isn’t the easiest to model accurately with atomistic force fields.
Cheers
Tom
Ask Google :)