No, I don’t. Try it and see.
Hi Dr. Lemkul,
So I tried performing both energy minimization and equilibration steps with the pull code in the .mdp files and then ran another 10-ns simulation with the pull code.
Unfortunately, it appears that the distance between the oxygen and Mg ion was even larger than in the previous simulation. I have attached a graph of the energy contribution from the pull restraint, showing it is far from 0 kJ/mol for almost the entire simulation after the starting coordinates.

I am not quite sure why this is happening. Should I maybe try increasing the strength of the pull restraint?
Thank you again for all of your help!
Is the distance correct after energy minimization? After equilibration? Are there other contacts that are interfering with the ability of the ligand and ion to be maintained at this distance?
The distance before energy minimization was 0.184 nm, and the distance after energy minimization was 0.208 nm. After NVT equilibration, the distance was 0.236 nm, and after NPT equilibration, the distance was 0.231 nm.
There are other contacts with the Mg ion, specifically two Glu residues, one Asp residue, and one to three water molecules depending on the simulation; the Glu and Asp residues change from a monodentate to a bidentate interaction with the Mg ion depending on the water molecules present.
Ideally, I would like to have both Glu residues and the Asp residue in monodentate interactions with the Mg ion, two water molecules coordinated to the Mg ion, and the oxygen of my ligand as the last interaction for the Mg ion.
So the situation is far more complicated than your original post indicated - I could have saved you a lot of time if your first post had all of the information that your most recent one did :) You don’t need to restrain a distance, you need to enforce a coordination geometry. Each of those distances should have a restraint on them (maybe not the waters, because additive force fields generally overestimate their affinity anyway).
I apologize, I should have given more information about my situation.
In order to enforce a coordination geometry, should I simply add more pull-coord-groups to the pull code?
In talking with my professor, I have added another restraint to the other oxygen of the ligand as well as the three restraints for the two Glu residues and one Asp residue.
Does the following pull code look like it will work properly?
; Pull code
pull = yes
pull-ngroups = 6
pull-ncoords = 5
pull-group1-name = a_10900
pull-group2-name = a_10909
pull-group3-name = a_10908
pull-group4-name = a_8361
pull-group5-name = a_8770
pull-group6-name = a_9183
pull-coord1-type = umbrella
pull-coord2-type = umbrella
pull-coord3-type = umbrella
pull-coord4-type = umbrella
pull-coord5-type = umbrella
pull-coord1-geometry = distance ; simple distance increase
pull-coord2-geometry = distance ; simple distance increase
pull-coord3-geometry = distance ; simple distance increase
pull-coord4-geometry = distance ; simple distance increase
pull-coord5-geometry = distance ; simple distance increase
pull-coord1-groups = 1 2
pull-coord2-groups = 1 3
pull-coord3-groups = 1 4
pull-coord4-groups = 1 5
pull-coord5-groups = 1 6
pull-coord1-dim = Y Y Y
pull-coord2-dim = Y Y Y
pull-coord3-dim = Y Y Y
pull-coord4-dim = Y Y Y
pull-coord5-dim = Y Y Y
pull-coord1-rate = 0.0 ; not pull, just distance restraint at a reference distance bw 2 groups
pull-coord2-rate = 0.0 ; not pull, just distance restraint at a reference distance bw 2 groups
pull-coord3-rate = 0.0 ; not pull, just distance restraint at a reference distance bw 2 groups
pull-coord4-rate = 0.0 ; not pull, just distance restraint at a reference distance bw 2 groups
pull-coord5-rate = 0.0 ; not pull, just distance restraint at a reference distance bw 2 groups
pull-coord1-k = 1000 ; kJ mol^-1 nm^-2
pull-coord2-k = 1000 ; kJ mol^-1 nm^-2
pull-coord3-k = 1000 ; kJ mol^-1 nm^-2
pull-coord4-k = 1000 ; kJ mol^-1 nm^-2
pull-coord5-k = 1000 ; kJ mol^-1 nm^-2
pull-coord1-init = 0.19
pull-coord2-init = 0.32
pull-coord3-init = 0.20
pull-coord4-init = 0.20
pull-coord5-init = 0.20
pull-nstxout = 0
pull-nstfout = 0
Thank you so much once again!
Looks reasonable. Try it and see!
I tried another 10-ns simulation with the above pull code in place. I also perfromed the energy minimization and equilibration steps for the 10-ns simulation with the pull code in place.
The vey beginning of the simulation was the best of everything thus far, but the ligand quickly drifted away from the desired distance from the Mg ion as the Asp residue and one of the Glu residues changed from monodentate to bidentate interactions with the Mg ion. Here is the energy output from the pull code:
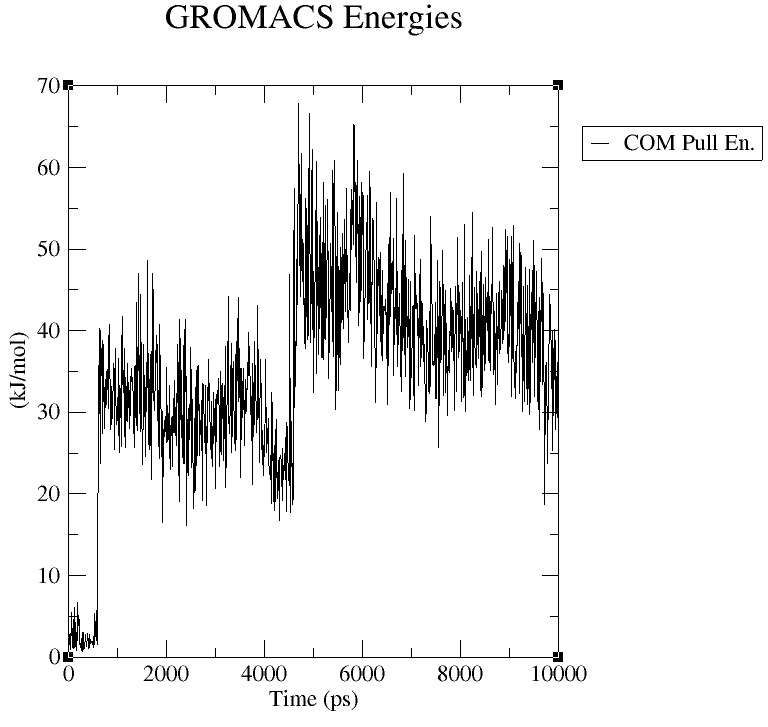
I am wondering if now I should try increasing the force constants for the restraints on the ligand oxygens? Also, would there be any benefit to using a ‘constraint’ pull-coord-type as opposed to an ‘umbrella’ pull-coord-type?
It’s worth a shot to increase the force constant, because you’re fighting against very strong forces that want to change the geometry. What you have to determine is whether the force field is actually producing a correct coordination and your assumptions are incorrect, or if the force field is somewhat imbalanced and you’re trying to “fix” the error by applying the bias. Dealing with divalent metals is challenging and most additive force fields model the interactions pretty crudely.
I wouldn’t advise using the constraint method, as that becomes very sensitive to changes because the distances are assumed to be fixed/rigid. With what you’re showing above, there are changes in geometry that would lead to constraint violations and the simulation would probably just crash.
I see. What would be an acceptable increase of the force constant to try out? Perhaps increasing it from 1000 to say 5000 kJ mol^-1 nm^-2?
Try it and see; I’m afraid there’s no real established rule here. You’re essentially fighting the magnitudes of interaction energies between the different species that want to drive the change in coordination.
Hi Dr. Lemkul,
I just wanted to follow up and let you know that the 5000 kJ mol^-1 nm^-2 force constant has worked in several simulations to keep the distance restraint in place.
Thank you very much for your help!
Josh
1 Like
Hi Josh.
I am also facing a similar situation.
My only confusion is that how you named the Mg and O of ligand as a_10908 and a_10900 respectively?
It seems that you have your gro file to have chain a to be already defined, which I do not have in my case.
FYI, I am attaching the link to my question in the grosmcs mailing forum: