I have done protein-ligand simulation for a protein and four different ligands. But all the pairs I have seen are leaving binding pocket immediately after the MDRUN had started.
Have pre-processed the trajetctory with “-pbc whole”, and “-pdb nojump”, and recentered using “-pbc mol -center -ur compact”, but the problem is not solved.
Ligand-protein-RMSD is behaving very weird for all the four ligands.
Below is a reference plot (from one of the ligand).
For protein-ligand complexes I always use the the following conversion:
get the starting conformation in pdb (with this you can have chain A, B etc in VMD visualization.
echo 1 1 1 | gmx trjconv -f traj.xtc -s sim.tpr -o conf.pdb -pbc cluster -center -boxcenter tric -e 0.001
convert the whole trajectory
echo 1 1 1 | gmx trjconv -f trtaj.xtc -s sim.tpr -o traj.xtc -pbc cluster -center -boxcenter tric (-dt X if you don’t want every snapshots)
If you still see your ligand dissociated then most likely your docking, even with the best score, gave false postive. That is why I say, that any docking pose needs to be confirmed for stability.
You most likely have a small molecule ligand, so you need to check how to make index groups and assign your ligand to one group and protein-ligand into another index group, let’s say group X and then replace in “echo 1 1 1” the ones to X. Later on in the pdb name protein as chain A and ligand chain B and you can do VMD… Generally you need to be familiar with the usage of index.ndx in gromacs
Hello @slovas
Thanks for your help.
I have created index group for ligand (Group 13), protein-ligand (Group 20) and run the following command,
echo 1 13 20 | gmx trjconv -f md_production.xtc -s md_production.tpr -o traj.xtc -pbc cluster -center -n index.ndx
And calculated the ligand rmsd using the following command,
gmx rms -s md_production.tpr -f traj.xtc -o rms-lig.xvg
Still, ligand RMSD is very high (aroubd 5-10 nm), graph looks almost identical to what I have attached before.
The box type I used is dodecahedron, and my protein is small (~ 200 residue). I will try running the simulation for cubic and triclinic box type, and see if the rmsd varies between them.
What surprised me the most is that this particular protein is showing a similar ligand-RMSD plot for all four ligands I tested. I ran simulations for other protein-ligand pairs using the exact same parameters and consistently obtained good RMSD values. So, I suspect that the volume of the dodecahedron box might be too large for the protein-ligand complex.
There is no such thing as a too big of a box, as far as I know. The reason to keep the box limited in size - in the limit of self interaction artifacts - is to limit the amount of solvent molecules in the box and, consequently, make simulations faster. There is also no post-processing data analysis that will fix the RMSD, apart from cleaning it up from any possible PBC artifact. The large RMSD is most likely because the ligand left the binding site, which you can check by visualizing the trajectory.
Yes, I agree obZehn, large box is not a problem. The opposite, however, is the truth. Using too small box one can introduce artificials in the simulations. For example, force the ligand to stay in the binding pocket. In fact, you should use the gmx mindist to check if the protein-ligand complex sees itself in the periodic box, which should not be the case.
Then I centered, made the complex compact by putting everything in a box, and finally fitted it using “-fit rot+trans”.
Upon visualizing the resultant trajectory along with the pbc box at different timepoints (0, 50,80,100), look like the ligand is not flying around, but the water molecules seem more defused than how it was before centering and fitting.
But RMSD is even worse than before.
So fitting and re-centering only made the waters more defused.
Below are the screenshot of the trajectory at different timepoints. final-100ns|402x500
What surprised me is, I selected “dodecahedron” for unit cell type, but the pbc box doesn’t look like one.
So, no matter what I try, RMSD is not improving, and although complex seems intact upon extracting the trajectory at different timepoints, the movie visualization seems the ligand moves a lot and far apart from the binding sites.
The simulation with cubic cell is not yet finished. I will update if there is any difference in RMSD.
Again, as long as you make sure that there are no PBC artifacts in the RMSD calculation, there is no post-processing that will make the RMSD better. If by visualizing the trajectory you see that the ligand left the pocket, then the ligand left the pocket and that’s it. You cannot fix this a posteriori, like lowering the RMSD or putting back the ligand in the pocket.
What do you mean with ‘complex seems intact’? Is the ligand still there there or not?
This is normal, you can probably recover the full box shape by adding the -ur compact flag to the trjconv command.
The blue line periodic box is just the way VMD handles dodecahedron, no worry about that. You should come to term with the fact that your ligand dissociated …
Even at the time-points where there are peak in the RMSD plot, the extracted trajectories look intact. the ligand is still there at the binding pocket.
OK, analyze rmsd of your protein and ligand separately. also analyze, for example, the distance between of center of mass of the inding pocket and center of mass of your ligand. Btw, most of the, not all, gmx analysis tools are PBC aware.
I have performed MD for few more ligands with this protein, and all the ligand RMSD are almost identical. Does that mean, this is not a good protein to do MD Simulation with these ligands?
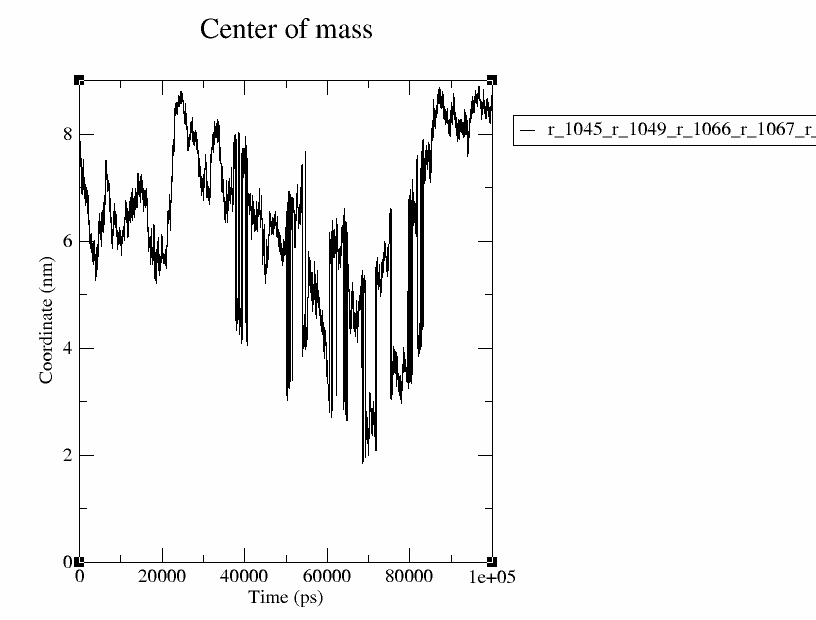
I have first made an index file for the interacting residues in the ligand binding domain (LBD) of the protein (P1045, L1049, P1066, Q1067, V1078, L1079, P1080), using the following command,
gmx make_ndx -f ../md_traj.gro -o LBD.ndx
And further the following when prompted,
r 1045 | r 1049 | r 1066 | r 1067 | r 1078 | r 1079 | r 1080
q
Next, I have made another index group with the LBD and the Ligand (group 13) using following command,
gmx make_ndx -f ../md_traj.gro -o LBD_lig.ndx -n LBD.ndx
And further typed following when prompted,
20 | 13
q
Next, I have calculated the distance between these two using gmx traj using the following command.









{kind=link}