GROMACS version:
GROMACS modification: Yes/No
Dear all
I want to simulate a Poly phenylene oxide system. In time steps of 1 fs I face with lincs warning(atom C and H rotates more than 30 degree). I changes frames to 1 ps to see what happens for C-H bonds in CH# group. in VMD I saw CH3 group rotates abnormally and I think this the reason that I encounter with lincs warning. Althought, in time steps of 0.5 fs I do not recieve such warning. I drew structure on the paper here to show the problem better. what should I do for that? What is the main problem.
Thank you
Can you provide an image of what you mean by this? It will be much more diagnostic than a generic hand drawing of the structure.
If you’re getting instabilities even at δt = 1 fs, it suggests your topology is not stable and you should check and refine your parameters.
Thank you for your reply
I checked the parameters related to these atoms. It was correct and their parameters are very well known. is it possible any problem in mdp file?
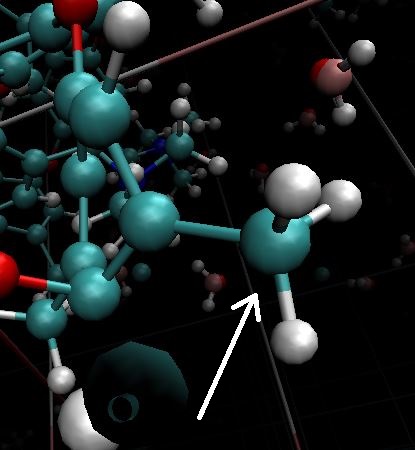
All problem I have is related to these C-H bonds
I don’t see anything wrong with that geometry. Do you have a snapshot of a distorted geometry?
Without seeing the .mdp file, I have no idea. Please upload it or copy and paste its contents.
Dear Lemkuls
Thank you for your guidance. this is my .mdp file for NVT
;title = BTMA in water (Rezayani)
;===========================================================
;define = -DDISRES (Nothing defined)
;disre = simple
;disre_weighting = conservative
;disre_fc = 10000
;===========================================================
; Run parameters
integrator = md
nsteps = 5000000 ; 5 ns
dt = 0.001
;==========================================================
; Output control
nstxout = 5000 ; save coordinates
nstvout = 5000 ; save velocities
nstenergy = 500 ; save energies
nstlog = 5000 ; update log file
nstxout-compressed = 500 ; xtc file
;===========================================================
; Bond parameters
continuation = no
constraint_algorithm = lincs ; shake is another one
constraints = h-bonds ; all-bonds can be used too
lincs_iter = 1
lincs_order = 4
;============================================================
; Nonbonded settings
cutoff-scheme = Verlet
ns_type = grid
nstlist = 10
rcoulomb = 1.0
rvdw = 0.9
DispCorr = EnerPres
;============================================================
; Electrostatics
coulombtype = PME
pme_order = 4
fourierspacing = 0.16
;=============================================================
; Temperature coupling is on
tcoupl = nose-hoover
tc-grps = system
tau_t = 1
ref_t = 295 ; based on article temeprature
;=============================================================
; Velocity generation
gen_vel = yes
gen_temp = 298
gen_seed = -1
;=============================================================
; Periodic boundary conditions
pbc = xyz
Nothing looks unusual, so I would revisit the implementation of the force field parameters to make sure they are correct and reproduce some sensible target data, like a QM potential energy scan.
Dear Lemkul
I checked my .itp file . I found the problem. Some bonds were written two times. I removed and correct repetitive bonds and the problem were fixed.
Thank you