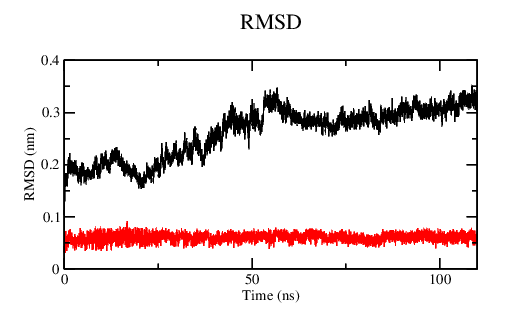
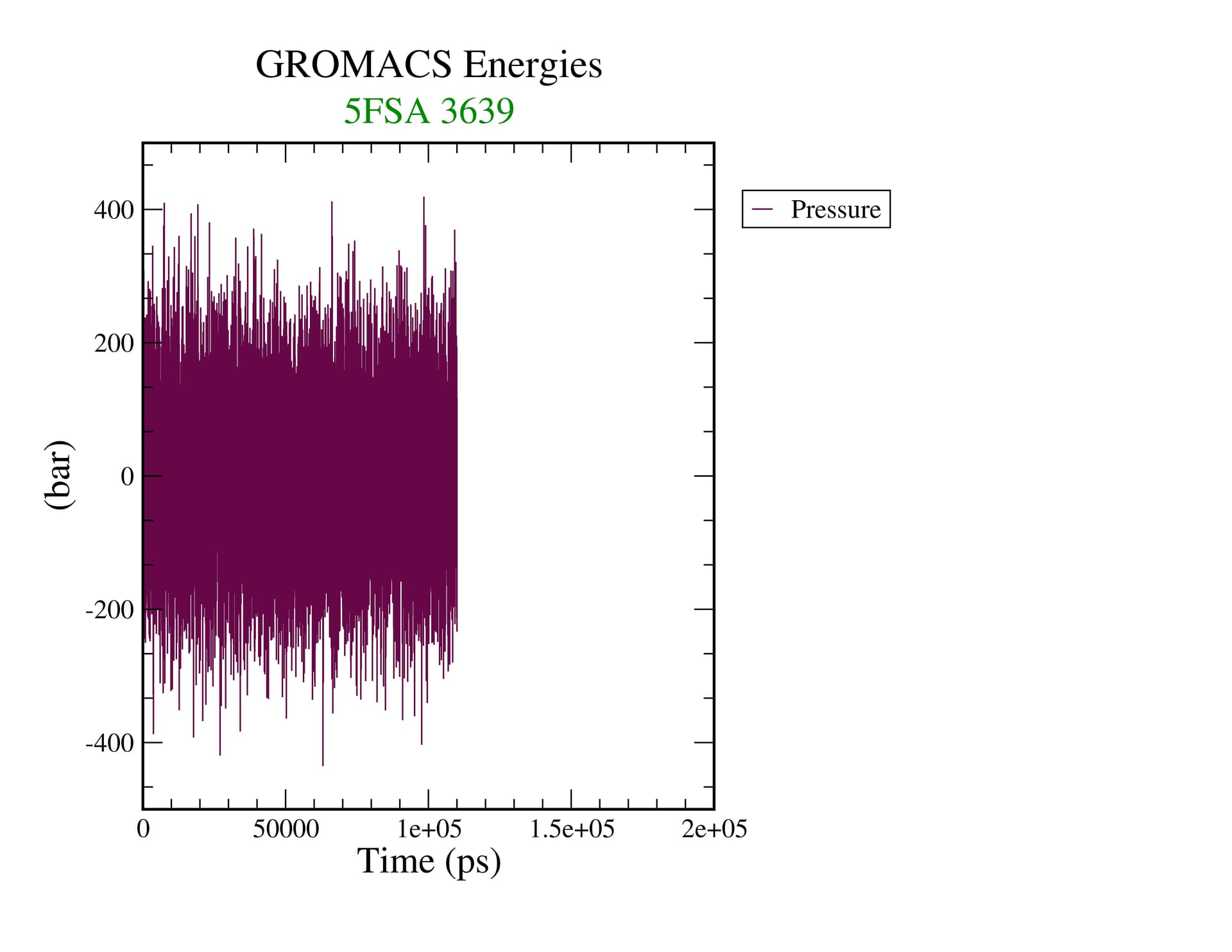
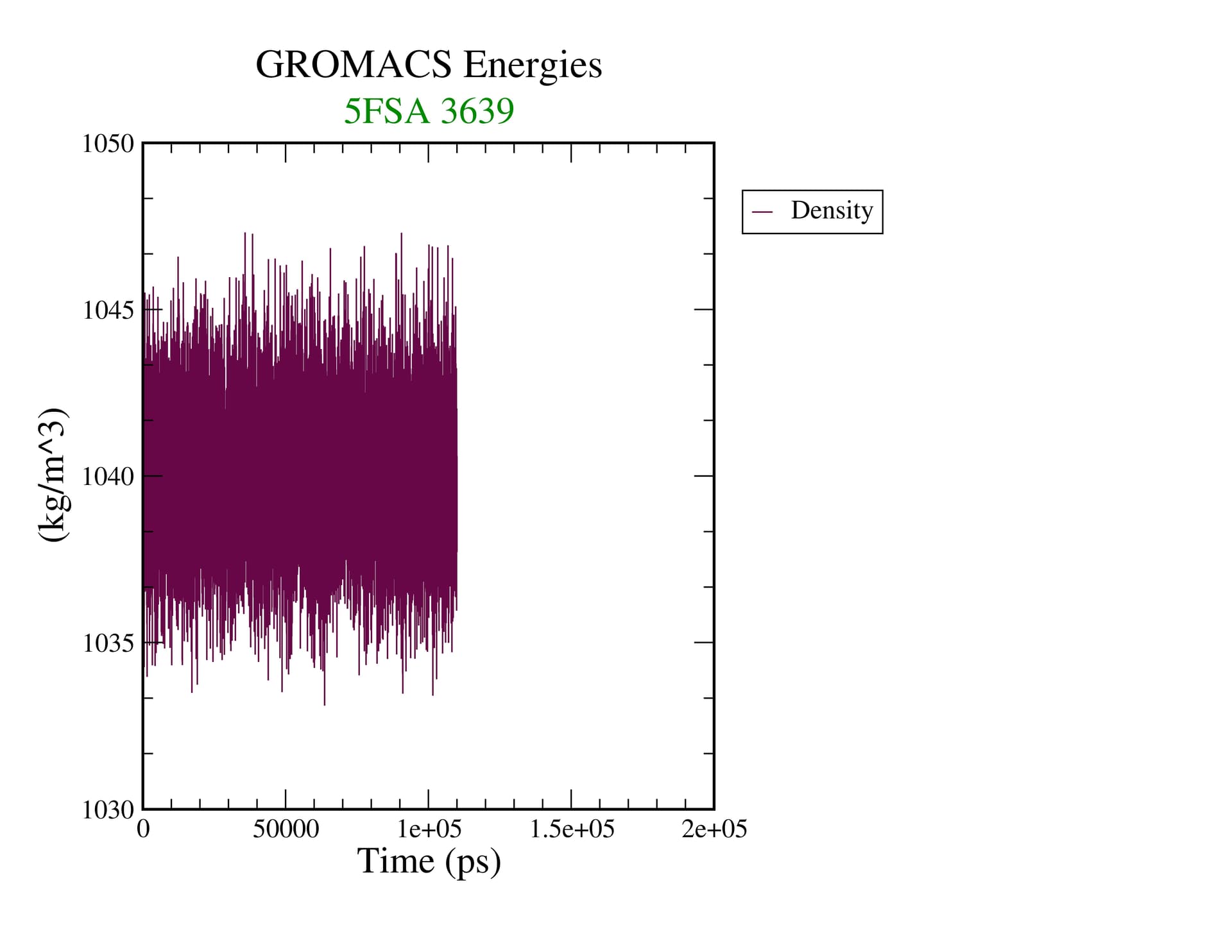
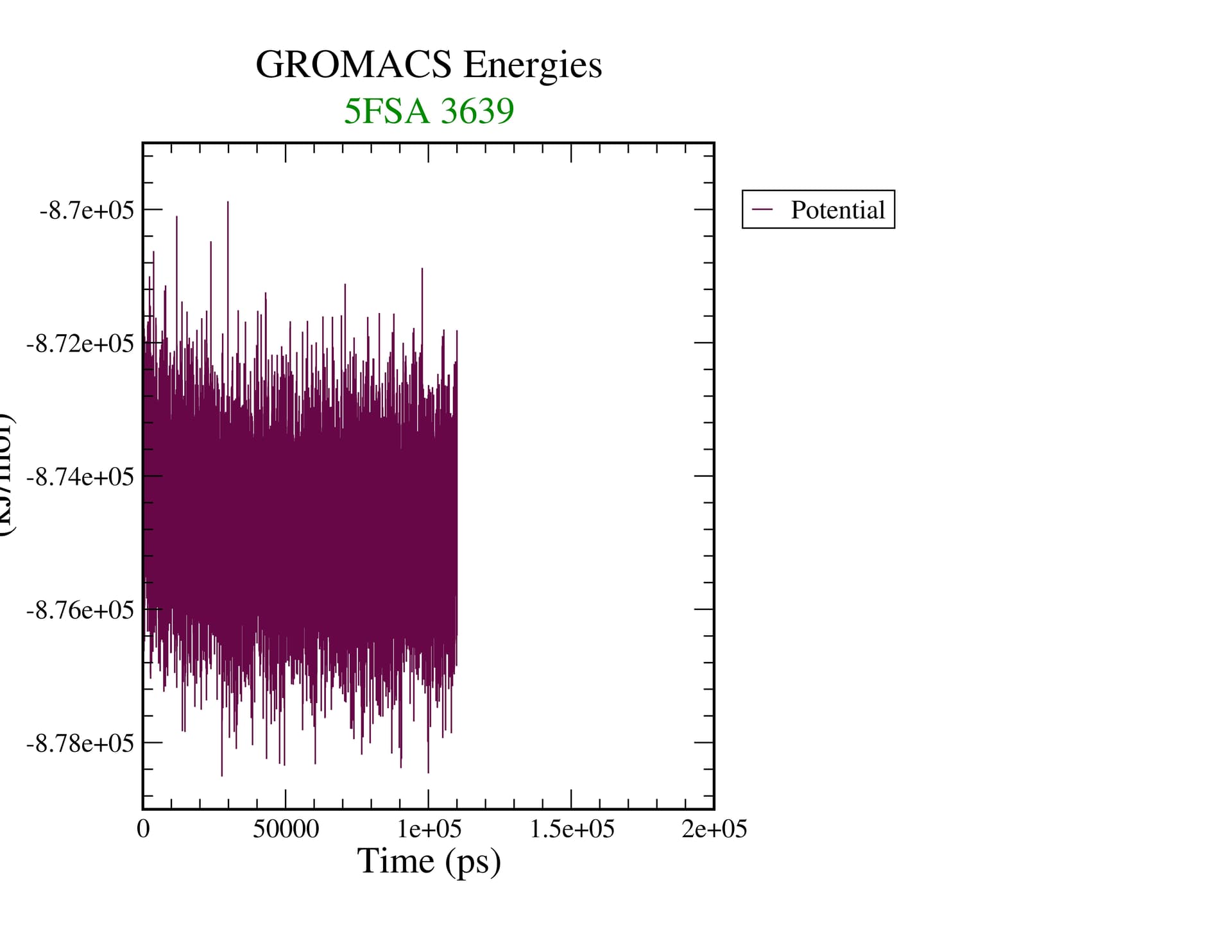
Equilibrium is a thermodynamic concept. RMSD doesn’t tell you anything about that. If you’re looking at assessing convergence, you have to be looking at all relevant structural, dynamic, and thermodynamic properties to see if they are statistically invariant over the time scale of your simulation.
I should be more specific. Equilibrium as a thermodynamic concept is derived from free energy of the system. The proxy for that in our simulations is whether or not relevant quantities in the ensemble are invariant. That means you have to assess the properties of interest in your system (structural, e.g. the conformations of your biomolecule(s)) and determine if you’ve achieved sufficient sampling. Nothing that gmx energy prints out tells you this, nor does the RMSD of the structure, as it is a degenerate metric (a single value can have many interpretations).