GROMACS version:2016.3
GROMACS modification: No
Here post your question
Hello,
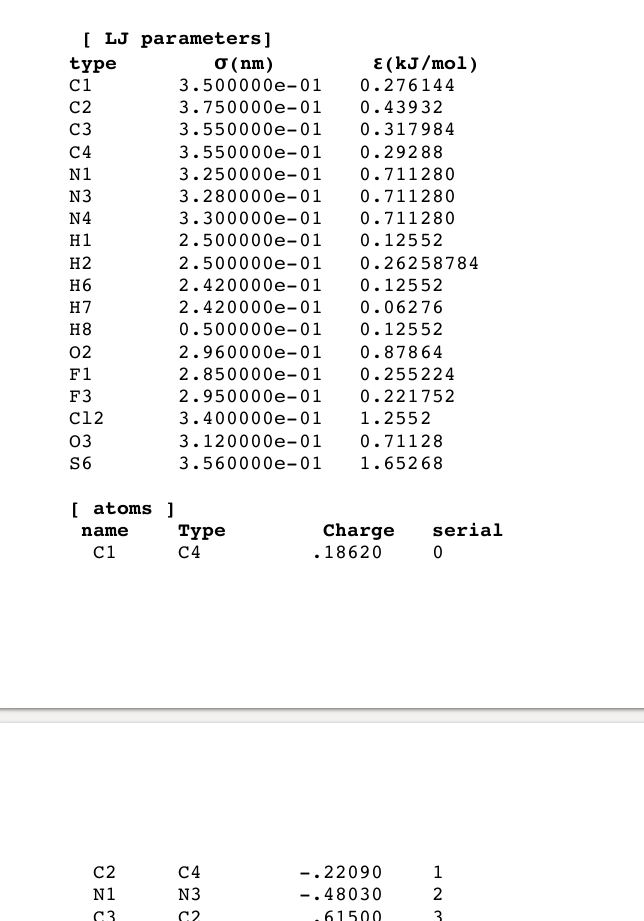
I wanted to model the drug-bound c-Src kinase with the dasatinib bound drug (RCSB: 3G5D) in GROMACS. For dasatinib parameters, I used the parameter values provided in Supplementary Information of “Role of Desolvation in Thermodynamics and Kinetics of Ligand Binding to a Kinase” (Figure attached). I have made an rtp file, and a modified pdb file (github gist) to name the atoms in dasatinib as per the parameters (residue name 1N1), which I made from the extent of my understanding in order to successfully build my system in gromacs. I wanted to use the OPLS-AA force field for the rest of the system.
Is this the correct methodology? How do I pass this rtp file to pdb2gmx, in order for it to pick up drug parameters?
Copied the rtp file to “/usr/local/share/gromacs/top/oplsaa.ff”, made an atp file containing atom types and their masses. (gist)
Ran pdb2gmx using OPLS-AA force field and TIP4P water.
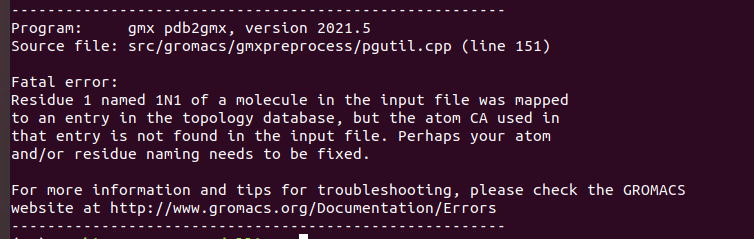
It gave me the following error :
Residue 1 named 1N1 of a molecule in the input file was mapped
to an entry in the topology database, but the atom CA used in
that entry is not found in the input file. Perhaps your atom
and/or residue naming needs to be fixed.
There is no CA entry in 1N1, residue database, or in the input file. How do I interpret this error?
Respected sir,
Thank you for your reply. I was not able to upload files earlier, hence I provided a link to a github gist in the previous post. Attached is the dasatinib.rtp file I made. I realize I did not provide the [bonded types] directive, but it assumed some type for now. I wish to understand how gromacs does the modelling, so I can provide the parameters in the right way. dasatinib_atp.dat (486 Bytes) dasatinib_rtp.dat (13.4 KB)
pdb file (final_drug_bound_1.pdb): final_drug_bound_1_pdb.dat (385.5 KB)
All the files are in dat format because of uploading constraints
Screenshot :
Please provide the full screen output from pdb2gmx. The error is not necessarily that informative. Everything that came before it is. There is a huge amount of diagnostic information that pdb2gmx prints that may be useful in pinpointing the problem.
Are you specifying 1N1 as Protein in residuetypes.dat? If so, don’t. pdb2gmx is trying to apply NH3+ and COO- patches to it as if it were a protein residue. That’s why you’re getting a fatal error.
In the future, please simply copy and paste the text rather than providing a series of screenshots with tiny text that is hard to read.
Respected Mr. Lemkul
Apologies for the format of the data.
I fixed the residuetype of 1N1 in residuetypes.dat to ‘Other’, and made some changes to the pdb. Attached is the pdb I now pass to pdb2gmx (GOL removed, TIP renamed to SOL) : final_drug_bound_1_pdb.dat (384.4 KB)
When I execute the same command, it doesn’t give an error on 1N1, but rather related to terminal residues, even after using the -ter option (GLY: 0 - NH3+, LEU: 0 - COO-) : pdb2gmx_out_dump.dat (10.7 KB)
I also tried modelling the terminal residues using CHARMM-GUI, but It provided an extra two atoms at the end of the protein, at the amino acid LEU, OT1 and OT2. I believe this is the COO- end for the protein. When I tried passing it through pdb2gmx, it said “OT1 was not found in residue LEU”, which is fair, considering the termini were added by CHARMM-GUI.
Kindly guide me as to how I should proceed. And thank you for your support so far.
The error has nothing to do with the termini, it’s incorrect nomenclature:
-------------------------------------------------------
Program: gmx pdb2gmx, version 2021.5
Source file: src/gromacs/gmxpreprocess/pdb2gmx.cpp (line 790)
Fatal error:
Atom OH2 in residue HO4 3 was not found in rtp entry HO4 with 4 atoms
while sorting atoms.
.
For more information and tips for troubleshooting, please check the GROMACS
website at http://www.gromacs.org/Documentation/Errors
-------------------------------------------------------
You’ve chosen TIP4P as your water model, which requires atoms OW, HW1, HW2, and MW. You’ve supplied atoms named OH2, H1, and H2 (e.g. CHARMM TIP3P nomenclature). Therefore, pdb2gmx fails to find the atoms it needs. I will also note that you may have issues with the topology you’re trying to generate anyway, because pdb2gmx will try to follow the .rtp entry for TIP4P (HO4), which does not specify a virtual site construction. You get this from tip4p.itp, but that means you should not include the water molecules in what you pass to pdb2gmx (though in this case, you have to generate the MW sites yourself).
Respected Mr. Lemkul,
Thank you for your input. I ended up removing the initial water molecules and solvating the system later. The pdb2gmx command ran successfully.
However, after specifying the box size, solvating the system (editconf, solvate), I ran grompp to generate a .tpr file, which gave me an error Atomtype C4 not found. Here is the output dump for grompp : grompp_dump.dat (3.1 KB)
I understand this error as the paper has introduced new atomtypes for the drug. When I searched online, it said I would have to make an itp file and specify (mass, sigma_LJ, epsilon_LJ and the charge) for each atom.
I have already provided LJ parameters in the rtp file, so is an itp file needed in this case? To phrase the question differently, how does one introduce a new atom type in a force field?
How can I estimate the charge value for each atom? That is not provided in the parameters of the paper. I could copy the charges from a different force field, but would that be the correct approach?
Also, how do I assign the chargegroup field for all atoms in the rtp file? I had allotted each atom its own chargegroup initially to see if gromacs accepted the file, which it did. But its not the right parameterization for the molecule.
There are no LJ parameters in .rtp files. You need to add them to ffnonbonded.itp under [atomtypes] or in an auxiliary .itp file in the system topology that defines the new parameters
Third time this week I’ve said this :) You cannot mix and match different force fields. A force field is a self-consistent entity and each has different parametrization strategies. If you need to develop parameters for any new species, you need to do some thorough reading about how to do this for your chosen force field. There are tools available for many force fields to aid in this process, but it is extremely odd to me that a published study provided new LJ parameters for something but not the charges. The two go hand in hand.
Charge groups are irrelevant in modern versions of GROMACS.
Respected Mr. Lemkul,
Thank you for your input. I generated an itp file and grompp worked. Apologies for stating that the charges weren’t present in the SI, because they were. In an itp file, each atom type is given a charge. But in the parameters provided, there were multiple atoms of 1 type with different charges, hence I took an arbitrary value of charge for the atom type and placed it in the itp file. I am banking on the fact that the charges specified in the rtp file override the ones in the itp file, as stated in the gromacs documentation.
I ran into another error during grompp : Proper Dih. multiplicity can not be perturbed grompp.
This error occurred because gromacs assumed I was providing parameters for proper dihedrals of the periodic type. I wanted to specify the use of Ryckaert-Bellemans dihedrals, which is function type 3. I did provide that in dasatinib.rtp and ran all of the steps again. I had to manually change the function from 1 to 3 in the itp file which gromacs generates after executing the pdb2gmx, editconf and solvate commands. How do I specify which functions to use without manually fiddling with the itp file generated by gromacs?
Nonetheless, I have successfully generated the gro and tpr files, and just need to verify if the system is modelled correctly. Thank you so much for your help and support.