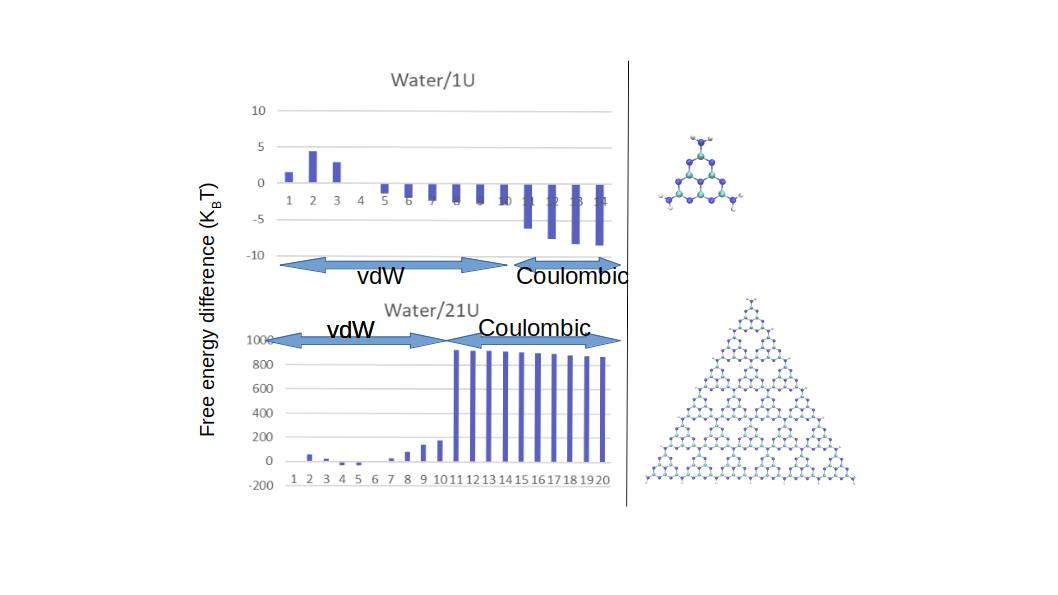
I have calculated the solvation free energy of two graphitic carbon nitride sheets (g-C3N4) of different sizes and the results are shown in the figure below.
My question is why for the smaller one the Coulombic interactions are negative while for the larger one they are extremely positive?
And, can we draw any conclusion based on the relative value of LJ and Coulombic interactions?
Lastly, the total values of solvation free energy in different solvents (water, DMSO, DMF, chlorobenzene) are almost the same for the larger sheet but they differ considerably for the smaller sheet. Why is that?
Without more information it is difficult to answer your questions. The different parts depend on the path you take and are therefore often difficult to interpret.
You don’t write how large your systems are and how long you simulate. But one explanation of the different results could be that your calculation are not converged and need to be run much longer.
Thank you Dr. Hess for your reply. I carried out a longer simulation (5 ns instead of 1 ns that I used to do) and the results were pretty as before (so for large sheet solvents has no effect on SFE). The paths I used were the same as follow:
The smaller sheet has 22 atoms and 2743 solvent molecules and larger sheet has 322 atoms and 7483 solvent molecules. SFE is negative for small sheet while it’s positive for large sheets.
I seems like something is wrong here. You should get attractive energies when turning on electrostatic interactions. What do you do with the sheet-sheet interactions? If you also perturb those, you need to do the same in vacuum.
The only difference between small and large sheets in the mdp file is this line:
couple-intramol = yes; for large sheet
couple-intramol = no; for small sheet
I also did not define any bonded-lambdas in the mdp file. The reason that I chose “yes” for the large sheet is that if I choose “no” for it I’ll get this error:
Fatal error:
There are 108 perturbed non-bonded pair interactions beyond the pair-list
cutoff of 1.23 nm, which is not supported. This can happen because the system
is unstable or because intra-molecular interactions at long distances are
excluded. If the latter is the case, you can try to increase nstlist or rlist
to avoid this.The error is likely triggered by the use of couple-intramol=no
and the maximal distance in the decoupled molecule exceeding rlist.
I tried increasing rlist but couldn’t solve the issue so I turned on the intramol.
The side length of the triangles (showing in the picture above) are 4.5 nm and 0.7 nm for the large and small sheets, respectively. Is there something wrong in my approach?
That explains it then. You are including the work to make the sheet disappear. When you use couple-intramol=yes, you need to run the same calculation without the water and subtract the free-energy difference you get from that.

{kind=link}