minim.mdp (1.2 KB)

GROMACS version: 2019
GROMACS modification: Yes/No NO
Here post your question
Hello all,
I am performing SMD-Umbrella Sampling for my micelle-peptide studies, based on Umbrella Sampling tutorial.
I performed 50ns simulation of Micelle-peptide-water system and took the micelle-peptide complex alone from the final snap for doing umbrella sampling.
GROMACS version 2019
FF: Charmm36 and water:TIP3P are used for all simulations.
Issue 1:
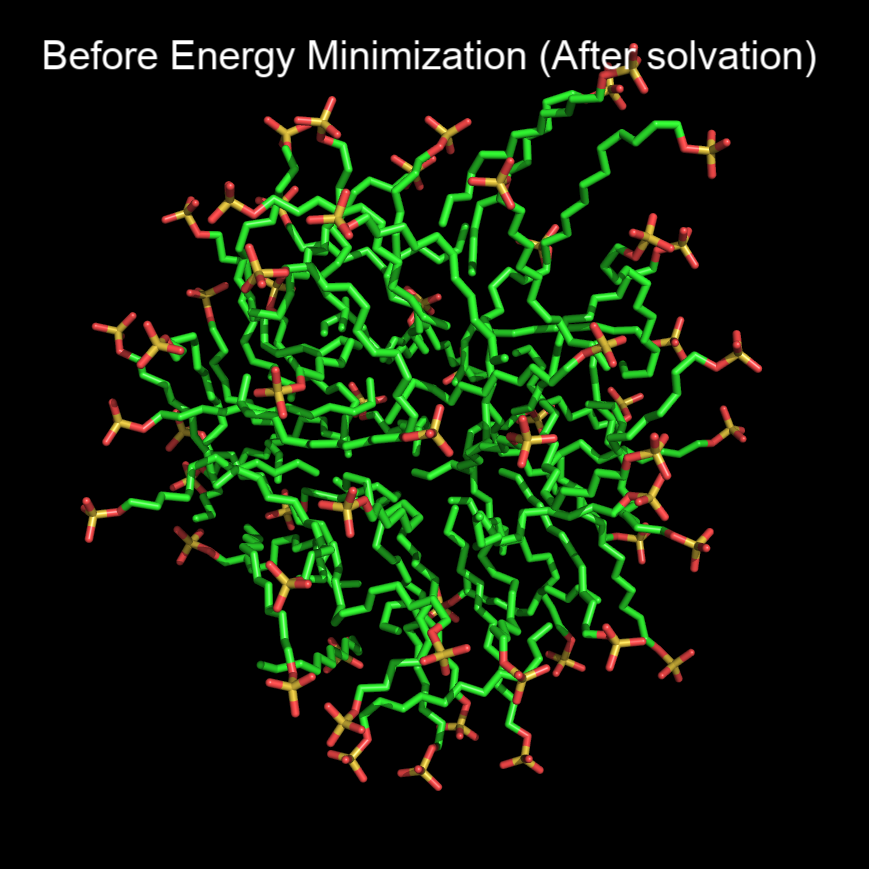
When i solvate the complex with new solvent molecules and performed energy minimization step. I saw, the hydrophobic tail of SDS molecules, which previously pointing towards micelle core are now exposed to water. Along with that, peptide’s secondary structure was completely destabilized to coils (peptide structure was so stable after the normal simulation [50ns] and during solvation step). So, I added restraints command in minim.mdp (attached below) with the hope that it will solve the issue but all i witnessed is, same buldging of SDS micelle with SDS molecules released from micelles (Images attached both before em and after em, unable to upload pdb/gro files) and destabilized peptide (peptide is not included as it is newly synthesized). I should not proceed with this deformed structures. How to preserve the structural integrity of original SDS and peptide?
Issue 2:
During MD pulling, we are restraining one group (using position restraints) and pulling the other group. Is it possible to provide soft restrain to the main chain heavy atoms or alpha carbons of the peptide with respect to center of mass of the same peptide [during pulling] separately (ie Peptide C-alpha with its same Peptide Center of mass)? (consider Protein in the place of peptide, if needed) if yes, how to acheive this? (The reason is to preserve 3D structure)
Thank you so much for reading this, kindly suggest me the ways to solve these two issues.