I am trying to run a pulling simulation on a long filament protein. I am using GROMACS pull code for this. The filament is about ~27 nm long. I set up a few residues on either side as Anchor and Pulled groups and pull corrd is set up between the two. The Anchor atoms are restrained using positional restraints. My goal is to do stretching simulations on the filament and look at its tensile properties.
I tried to use the pull_coord_geometry as distance, but it gives me a fatal error saying the distance between pulling group is larger than 0.49 times the box size. My box size is 60x20x20 (triclinic). The first dimension is the one parallel to the filament axis. I aligned the filament to the X-axis during the system preperation.
I also tried using direction/direction-periodic as the geometry. I tried with both a 1 0 0 vector and a manual vector connecting the Anchor and Pulled COMs. In both cases, my filament just bends (with the Anchor and Pulled group coming close to each other). I tried different spring constants and velocity values, but the same happens in all the cases. I also tried making my Pulled group just a single atom (the CA of the residue at the tip), but it did not help.
I am attaching a represtantive pull.mdp file below.
It would be great if anyone has any insight into why the above problem is happening, and if there is any solution.
Note: I am currently trying to run the simulation with a dodecahedron box of 60x60x60, so that I can use the distance geometry. But I feel like this is an overkill with the number of solvent molecules I am going to have in this setup.
I don’t see any mistakes here, apart from the force constant being quite low. I think this should work.
But note that your pull rate is very high. I don’t think you would get useful results when you get this to work. Such SMD simulations require far more simulation time than you can achieve.
I had lowered the force constant to see if the snapping back/bending of the filament is due to high force constant. I think I even tried as high as 1000.
The pull rate itself - I don’t really know what value I need. I just tried this out to see if it’s behaving properly, so that I can play around with the values later. But I can’t even get it to work as expected.
In my case, instead of the Pulled group moving away from the Anchor, it just snaps towards the Anchor in the first few picoseconds of the simulation. Do you think any of my paramters could be the reason for this behaviour?
I’m not entirely sure what the issue is that would cause the error but I noticed a couple of things in your post, please correct me if I’m interpreting them wrong.
In the comments you have “~5 nm at 1 nm ns⁻¹” and “1 nm ns⁻¹”. Was that the original pulling rate and duration you were using with the distance geometry? Since your protein is 27 nm long, that error might trigger after ~3 ns with a 1 nm/ns pull rate because of the period boundary conditions. However, if the error occurs on shorter simulations or with smaller pulling rates you shouldn’t be reaching that limit. I’m not sure a “dodecahedron box of 60x60x60” is what you need to resolve the issue with the distance geometry. If the issue is the PBC, if you stick with the triclinic box, but change it to 65 or 70 in the x direction that might correct the issue.
You also mentioned when using the direction/direct-periodic geometry that the filament bends instead of stretching. This sounds like you may have two groups reversed, such that x_Anchored(t=0) > x_Pulled(t=0). So what might be happening is you are pushing the “pulled” group towards the anchored group. Have you tried switching the direction of the vector (pull-coord1-vec = -1 0 0) with this geometry?
I’m not sure if this last point would have any impact on your simulation. The pull-coord1-dim variable is set to act in all directions by default. As you are only acting in the X direction you could set pull-coord1-dim = Y N N.
Good point about the switched order/direction. Sign issues are common and tricky.
This is precisely the type of case for which I designed direction-periodic. But I would think that in your case you can easily run into issue of the molecule interacting with its periodic images.
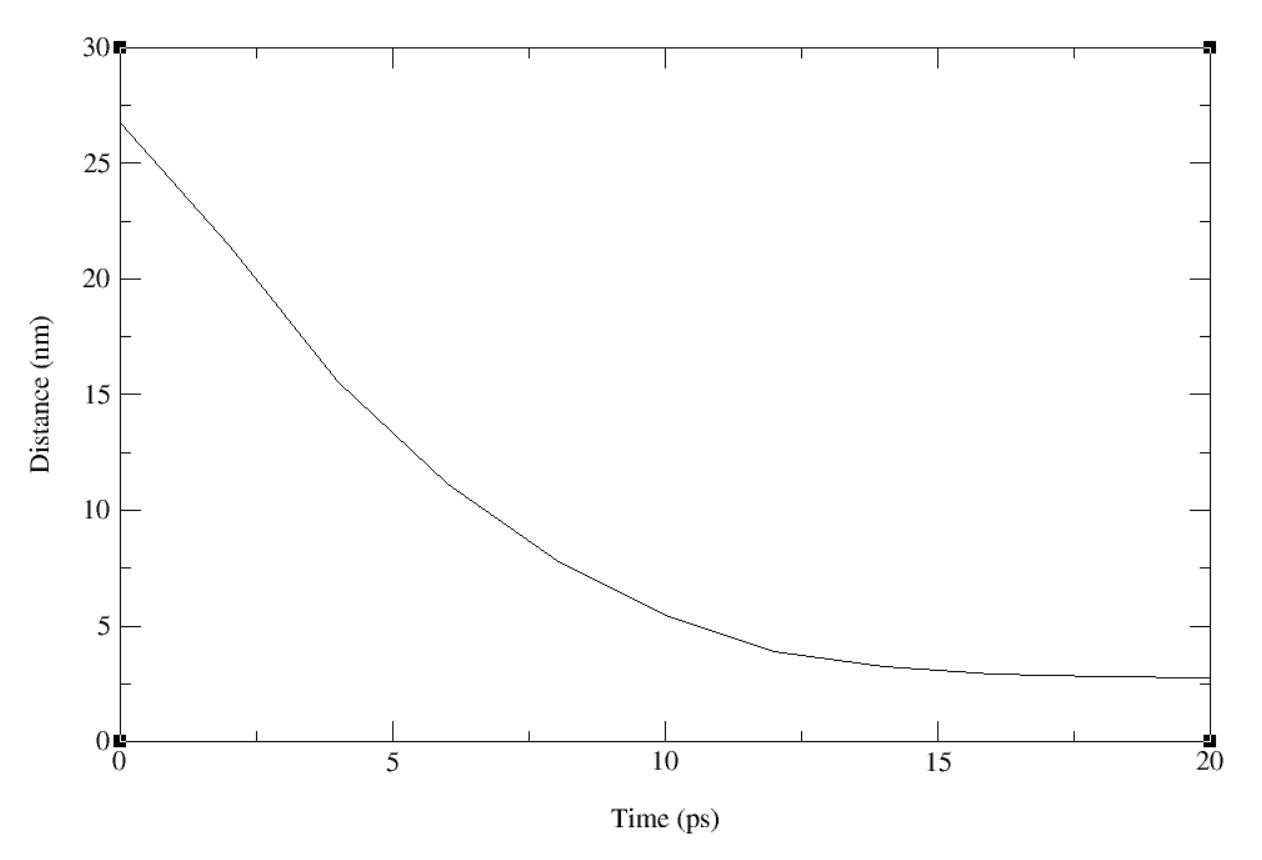
Regarding the periodic boundary conditions, I did try with smaller pull rates, and the bending still happens, and it happens in the first few picoseconds. I tired these with the direction-periodic geometry. When I say bending, what really happens is the residues in the Pulled group (~15 residues) move towards the Anchor group, unfloding the subunit containing the Pulled group residues and causing a bending movement. I am attaching an image of the distance plot between the Anchor and Pulled groups. Different pulling rates give me similar plots with different decay rates.
Interestingly, the same behaviour happens even when I don’t have any pulling rate (zero pulling rate) and just a spring constant.
I also tried a bigger dodecahedron box (60x60x60) and distance geometry, but that also gave me a similar bending behavior. Regarding bigger boxes in the x-direction, I tried that with keeping the other dimensions as 20 (My filament is about 10 nm in width). But since the distance between Pulled and Anchor group (~27 nm) is bigger than 0.49 times the smallest box edge (10 nm), GROMACS does not allow me to use distance geometry.
I also tried switching the signs in the pull vector, it does not seem to have made any difference. I am currently trying a slightly bigger triclinic box (70x30x30) and direction-periodic geometry to completely rule out any PBC issues. I am not sure if its going to help though.
Please let me know if you have any suggestions based on these information.
Update:I was able to get it to work as expected by adding a reference distance using pull-coord1-init = 26.681. I think it works as expected now.
Also, @hess , you mentioned in one of your previous replies that the pull rate is high and I might not get useful results from it. Could you please explain what you meant by that? In my case, I am trying to compare different filament with different subunit interfaces and compare their relative strength and things like that. Does this appraoch with a stretching simualtion make sense in this case? Did you mean that I have to just lower the pull rate or change the appraoch completely?
I was about to ask whether you have pull-coord1-start set to yes, that would also solve the issue.
My guess is that you end up pumping thousands of kJ/mol of energy into your system by pulling with that rate. That means that you might not at all be sampling conformations that you would sample at much lower pulling rates. You can compute the work on the system by integrating the force curve and multiplying by the pull rate.