GROMACS version: 2019.3/intel-2018
GROMACS modification: No
Hi,
I’m trying to do a simple pulling simulation in the z direction. I’ve decided to put a position restraint on the N terminus of my protein (the entire first residue) and pull on the C terminus (the full last residue). Here is my pull code.
; Pull code
pull = yes
pull_ncoords = 1 ; only one reaction coordinate
pull_ngroups = 2 ; two groups defining one reaction coordinate
pull_group1_name = Chain_A
pull_group2_name = Chain_B
pull_coord1_type = umbrella ; harmonic potential
pull_coord1_geometry = distance ; simple distance increase
pull_coord1_dim = N N Y
pull_coord1_groups = 1 2
pull_coord1_start = yes ; define initial COM distance > 0
pull_coord1_rate = 0.001 ; 0.01 nm per ps = 10 nm per ns
pull_coord1_k = 1000 ; kJ mol^-1 nm^-2
pull_group1_pbcatom = 1957 (mid atom number of the last residue)
pull_pbc_ref_prev_step_com = yes
Looking at my pullf.xvg file, I can’t interpret where I’ve gone wrong.
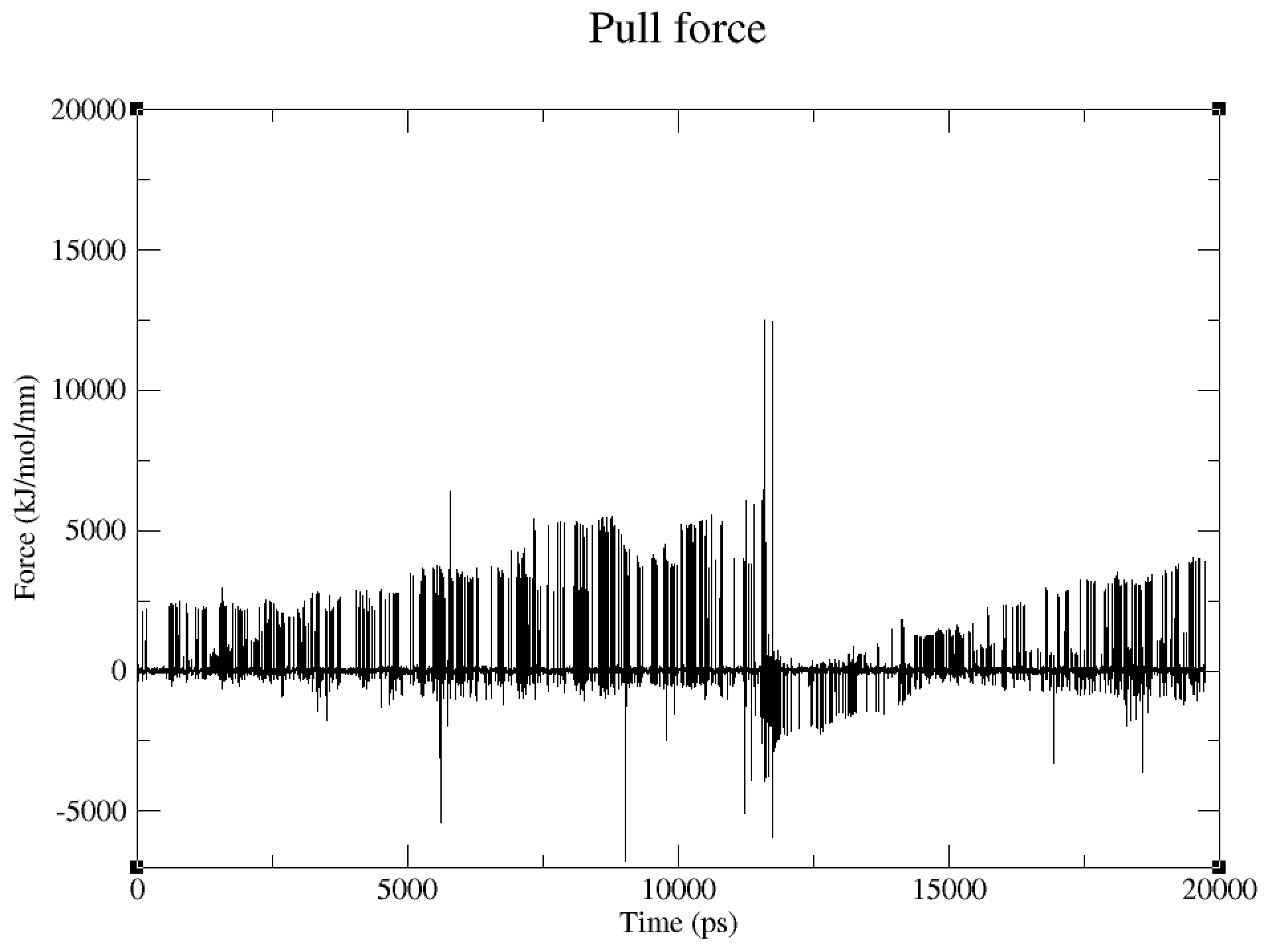
Looking at my trajectory in VMD, it looks like both ends of my protein is being pulled?
