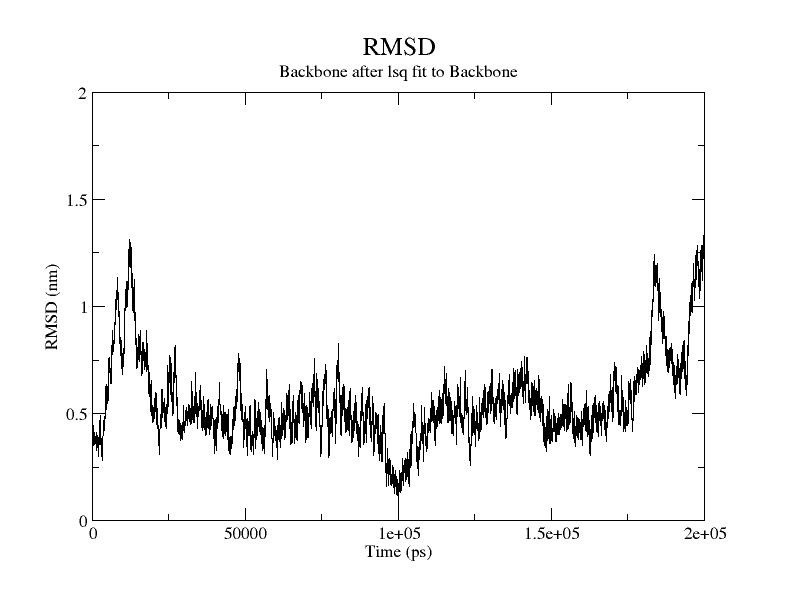
I guess this is an RMSD plot of the backbone of the whole structure, so both proteins together?
First, I wouldn’t say that for the first 250ns the system is stable. Already after 15ns or so you have large fluctuations up to ~1.3 nm, and same towards the end of the 250ns, which in principle are quite large numbers. The fluctuations are telling you that the system is re-organizing itself a lot, and most likely detaching and reorienting, although it’s hard to state anything as the RMSD is an extremely degenerate metric. One of the things that should be done here is check the trajectory, and as you said you see the system detaching and flying apart, so you have a reason for these numbers: the dimer doesn’t stick together.
Is this normal? the production run trajectory motions also showed unbound state over 500ns run. is this normal? or i have to apply gentle force to the system to keep it together? Whats the point of the complex if it drifts apart and stays independent?
I am unable to understand what to do? should i go for fresh run or any other possible solution? I need suggestions on this.
These are questions for you, as your simulations will be based on your scientific inquiry. Some proteins interact and stick together, some other don’t, there is no normal or expected behavior, and there is no general solution. Applying a force to keep them together will solve the problem of them detaching, but you need a justification. Enforcing, for example, a wrong interface will give just garbage as output results. Personally, I would check
i) if the force field is suitable for this complex. Am I using a ff that is notoriously bad at reproducing the quantities I’m looking for?
ii) What has been done previously in literature? Did someone simulate my complex? What problems do they have?
iii) my starting structure. Is this a crystal? Is this a complete structure or am I neglecting part of the proteins that can stabilize the interactions, like a membrane domain or an interacting loop?
iv) my equilibration protocol. Am I being too brutal? Maybe a slow warm up of the system with hard position restraints is needed.
v) run more than one replica. Nowadays, any result should not be presented as a single replica, but should be repeated a few times, especially for unbiased sims.