GROMACS version: 2022.1
GROMACS modification: No
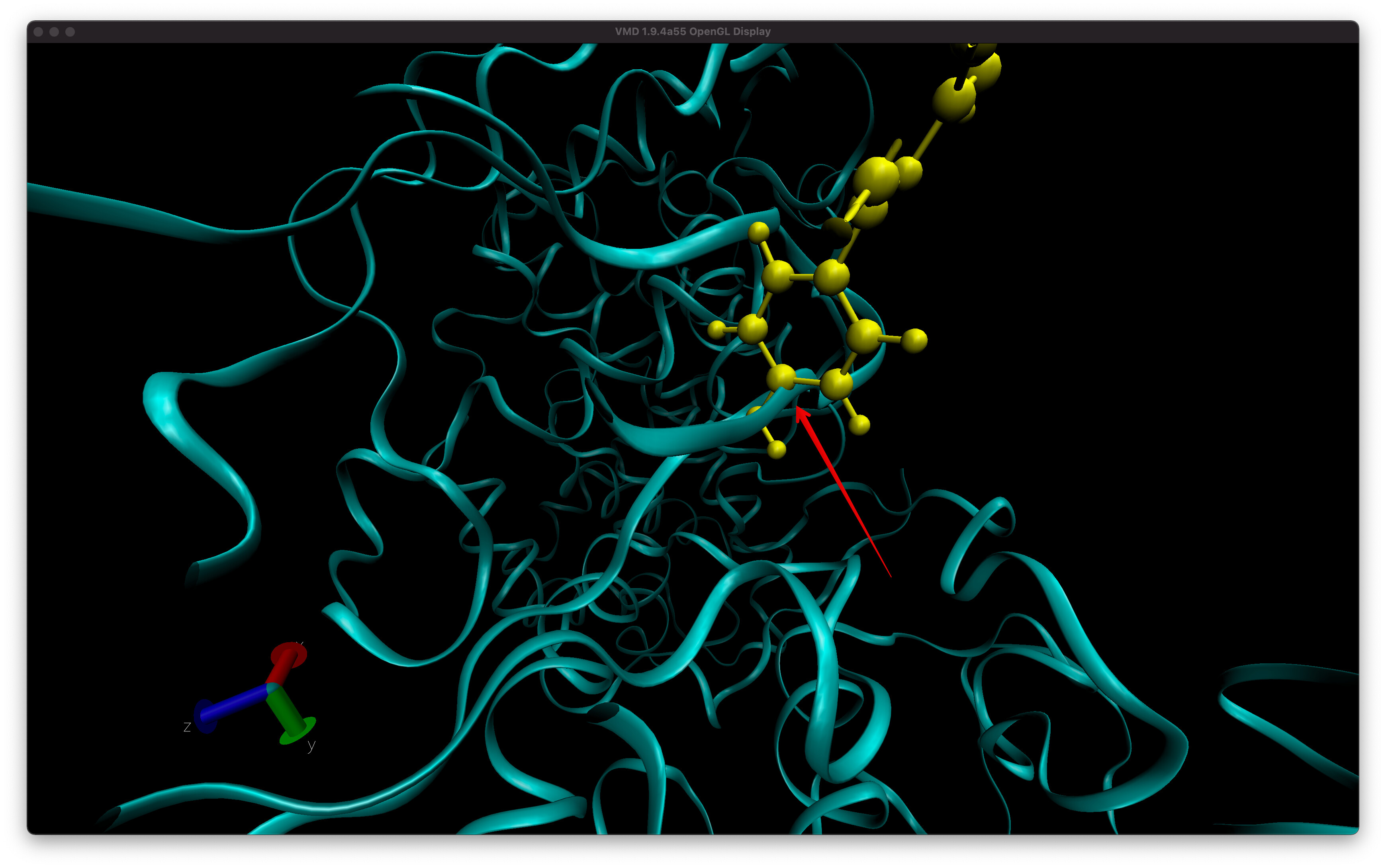
Why do I see overlapping of atoms in VMD for my trajectory in production dynamics. I ran all the minimization and equilibration already. The first screenshot is from frame 15 and there is overlap between ligand and backbone atoms. You see this with CPK representation as well but harder to visualize. The second images is the equilibrated starting structure and there is no overlap. How can I confirm from a snapshot, i.e. frame 15 if there is indeed overlap of atoms?
I converted trajectory and .gro file with below commands and select non-water for both.
gmx trjconv -f CompE1_V1.xtc -o complex.xtc -s CompE1_V1.tpr -center -pbc nojump -ur
compact
gmx trjconv -f CompE1_V1.gro -o complex.gro -s CompE1_V1.tpr
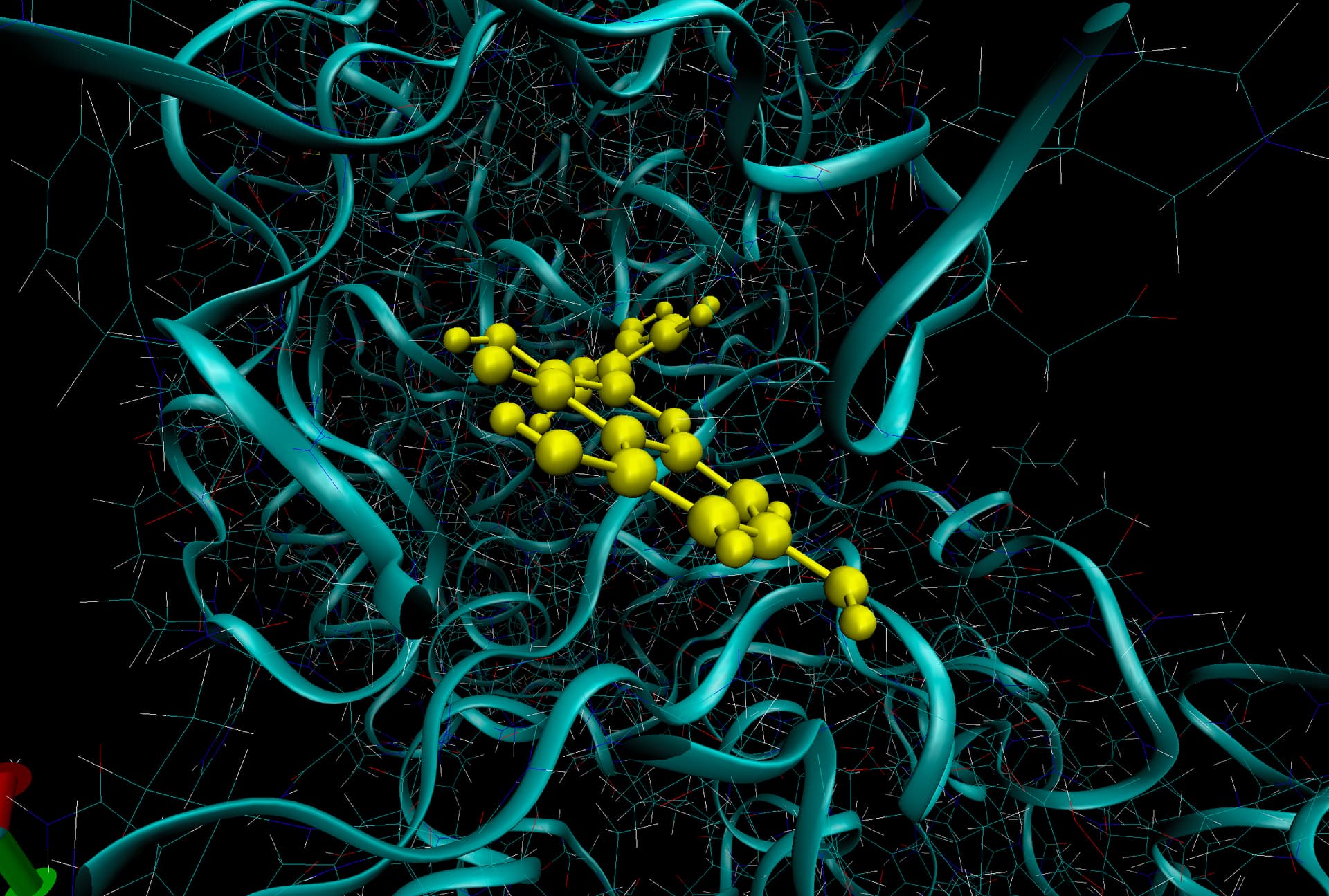
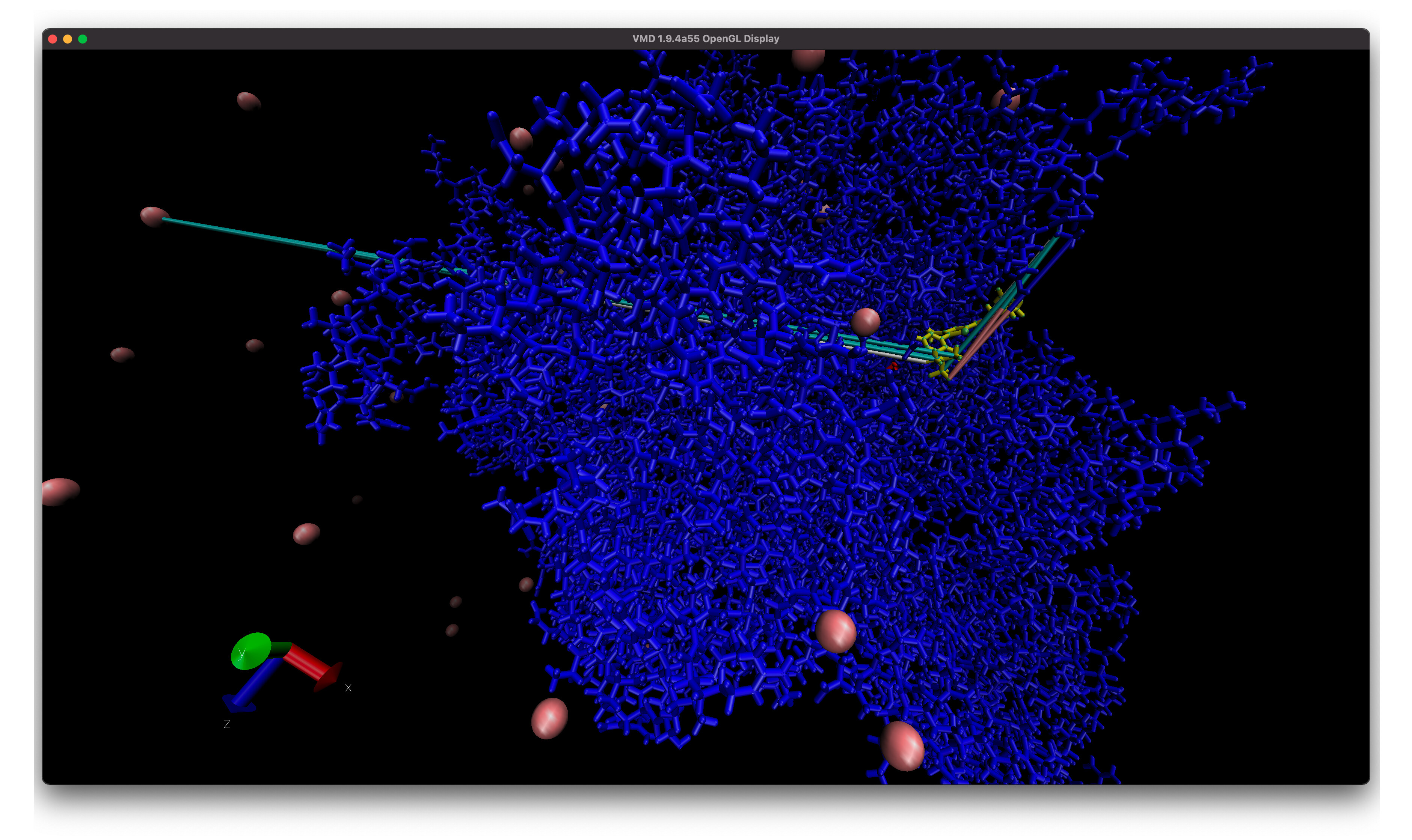
I also have another problem with a different trajectory and system with some some atoms (I think) near the ligand have really long lines in VMD and they seem to start near the ligand and end at a CL ion (left arrow) or some random other set of protein atoms (right arrow). And if I add water to the visualization there are more of these long lines! Is this more of an issue with VMD? I is much easier to see issue when I add bonds to the entire system (second image).
For the first issue on clashes, you should be able to simply issue gmx trjconv -pbc mol -center and center on the protein. Everything else will fall into place.
The second issue of wild lines just means you haven’t made molecules whole appropriately. gmx trjconv -pbc mol should fix this easily.
I seem to always see the ligand clashing with the protein no matter which way I run trjconv either with -pbc mol, -pbc nojump, -pbc whole, all with -center and choosing to center either on protein, protein and ligand, non-water. Tried it all. Seems very odd this is occurring.
Actually now that I am looking at a different lambda state of coul-lambda=0.0 and vdw-lambda=0.0 (this analysis was before I knew we should be running MM-PBSA vs. lambda states for FEP for large biomolecules from Justin’s other post) it seems to be fine. I was actually concerned when I saw my ligand flying all over the place in the coul-lambda=1.0 and vdw-lambda=1.0 state. Does this mean if I already ran my simulations I should use the coul-lambda=0.0 and vdw-lambda=0.0 state simulations for MM-PBSA? Or should I rerun completely without free-energy = yes?